Effect of Dexmedetomidine on Propofol-induced Neuroapoptosis by Reducing the Mitochondrial Membrane Potential in Primary Cultured Cortical Neurons of Rat
李建立1,, 郭洪霞1, 梁巍2, 容俊芳1, 刘锐3,
1.河北省人民医院麻醉科,石家庄 050051
2.河北省人民医院普外科,石家庄 050051
3.河北省胸科医院,石家庄 050041
LI Jianli1,, GUO Hongxia1, LIANG Wei2, RONG Junfang1, LIU Rui3,
1.Department of Anesthesiology,Hebei General Hospital,Shijiazhuang 050051,China
2.Department of General Surgery,Hebei General Hospital,Shijiazhuang 050051,China
ObjectiveTo investigate the effects and mechanism of dexmedetomidine on propofol-induced neuroapoptosis in primary cultured cortical neurons. MethodsAfter the neurons being cultured for 7 days,they were divided into vehicle control group (equal volume of 20% fat emulsion),propofol group(500 μmol·L-1) and dexmedetomidine+propofol group (dexmedetomidine at 0.001,0.01,0.1 1 μmol·L-1and propofol at 500 μmol·L-1).Twelve hours after treatments,neuron viability was measured by MTT assay,neuron structure was analyzed by microscope.Neuroapoptosis was detected by Hoechst33258 staining and mitochondrial membrane potential was measured by the fluorescent dye rhodamine 123 (Rh123). ResultsCompared with the vehicle control group,propofol inhibited neuron viability greatly(P<0.05).Compared with propofol treatment group,dexmedetomidine increased neuron viability in a dose-dependent manner (P<0.01).Lack of three-dimensional sense,faded color and unclear outline were observed,fractured neuron axons or neurons death were also observed in neurons treated by 500 μmol·L-1 propofol.While dexmedetomidine inhibited propofol-induced morphological damage,propofol (500 μmol·L-1) markedly increased the number of apoptotic neurons (P<0.01) and decreased the mitochondrial membrane potential greatly (P<0.01).Dexmedetomidine (0.1 μmol·L-1) significantly decreased the number of apoptotic neurons (P<0.01) and increased the mitochondrial membrane potential (P<0.01). ConclusionDexmedetomidine exerts its neuroprotective effects against propfol-induced neuroapoptosis by protecting the mitochondrial membrane potential.
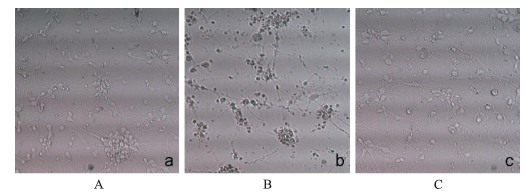
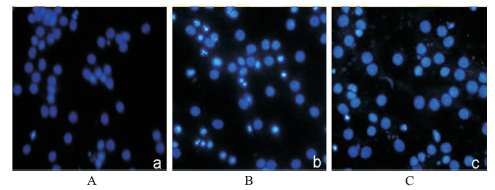
Fig.2
Effect of dexmedetomidine treatment on propofol-induced neuroapoptosis in primary cultured cortical neurons(×200) A.control group;B.propofol group;C.dexmedetomidine plus propofol group
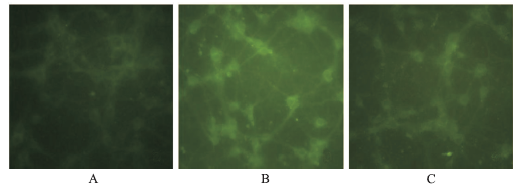
Fig.3
Effect of different treatments on mitochondrial membrane potential of primary cultured cortical neurons(×200) A.control group;B.propofol group;C.dexmedetomidine plus propofol group
丙泊酚通过激动γ氨基丁酸A型受体(Gamma amino acid type A receptor,GABAR)和抑制N-甲基-D-天冬氨酸受体(N-methyl-D-aspartic acid receptor,NMDAR)发挥麻醉作用,《中华人民共和国药典》2010年版提示丙泊酚慎用于<3岁患儿,但该药具有起效快、苏醒快、并发症少等优点,在实际临床工作中仍被广泛应用于婴幼儿麻醉的诱导与维持。近年来大量动物实验和细胞研究表明,丙泊酚对发育期大脑具有神经毒性[1-4],因此丙泊酚临床应用,尤其是在婴幼儿麻醉中的应用,引起广泛关注。本研究发现,丙泊酚可对原代培养皮质神经元产生损伤,表现为神经元形态学损伤,细胞存活率下降,细胞凋亡率上升,出现细胞核固缩和裂解等细胞凋亡的典型表现。
针对麻醉药引起的发育期大脑损伤,美国国立卫生研究院(national institutes of health,NIH)、美国食品药品管理局(food and drug administration,FDA) 以及国际麻醉研究学会(international anesthesia research society,IARS)要求不仅要研究麻醉药神经毒性的发生机制,而且要寻找有效的措施防治麻醉药引起的发育期神经损伤。众多学者对如何防治丙泊酚对婴幼儿大脑产生的神经损伤进行了广泛研究,发现锂剂、乙酰左旋肉碱等对丙泊酚引起的神经元损伤有保护作用[7-8],但它们在婴幼儿使用的安全性有待进一步探讨。盐酸右美托咪定是另一高选择性α2肾上腺素受体激动药,具有镇静镇痛作用,作为麻醉辅助药物广泛应用于临床麻醉。目前大量研究发现,盐酸右美托咪定具有神经保护作用,如对缺血-再灌注损伤[9],高氧诱导的发育期大脑损伤[10]产生保护作用。另有动物实验研究表明,盐酸右美托咪定可对麻醉药氯胺酮、异氟烷以及丙泊酚引起的发育期大鼠大脑凋亡样损伤产生保护作用,但盐酸右美托咪定减轻麻醉药发育期神经毒性的机制目前还不清楚[6,11-12] 。另有研究认为,盐酸右美托咪定本身对发育期大脑不产生神经损伤作用[13]。盐酸右美托咪定是否对抗丙泊酚诱导的原代培养皮质神经元损伤,笔者未见报道,故在本实验中对此进行了研究。
HUNJ,JINS,CHENX,et al.Propofol administration during early postnatal life suppresses hippocampal neurogenesis[J].Mol Neurobiol,2016,53(2): 1031-1034.
Propofol is currently one of the most widely used intravenous anesthetics and has been indicated to induce cognitive dysfunction in adults. Here, we investigated the effects of propofol exposure durin
KARENT,SCHLAGER GW,BENDIXI,et al.Effect of propofol in the immature rat brain on short-and long-term neurodevelopmental outcome[J].PLoS One,2013,8(5):e64480.
Background Propofol is commonly used as sedative in newborns and children. Recent experimental studies led to contradictory results, revealing neurodegenerative or neuroprotective properties of propofol on the developing brain. We investigated neurodevelopmental short- and long-term effects of neonatal propofol treatment. Methods 6-day-old Wistar rats (P6), randomised in two groups, received repeated intraperitoneal injections (0, 90, 180 min) of 30 mg/kg propofol or normal saline and sacrificed 6, 12 and 24 hrs following the first injection. Cortical and thalamic areas were analysed by Western blot and quantitative real-time PCR (qRT-PCR) for expression of apoptotic and neurotrophin-dependent signalling pathways. Long-term effects were assessed by Open-field and Novel-Object-Recognition at P30 and P120. Results Western blot analyses revealed a transient increase of activated caspase-3 in cortical, and a reduction of active mitogen-activated protein kinases (ERK1/2, AKT) in cortical and thalamic areas. qRT-PCR analyses showed a down-regulation of neurotrophic factors (BDNF, NGF, NT-3) in cortical and thalamic regions. Minor impairment in locomotive activity was observed in propofol treated adolescent animals at P30. Memory or anxiety were not impaired at any time point. Conclusion Exposing the neonatal rat brain to propofol induces acute neurotrophic imbalance and neuroapoptosis in a region- and time-specific manner and minor behavioural changes in adolescent animals.
ZHONGY,LIANGY,CHENJ,et al.Propofol inhibits proliferation and induces neuroapoptosis of hippocampal neurons in vitro via downregulation of NF-κB p65 and Bcl-2 and upregulation of caspase-3[J].Cell Biochem Funct,2014,32(8):720-729.
Propofol is widely used in paediatric anaesthesia and intensive care unit because of its essentially short-acting anaesthetic effect. Recent data have shown that propofol induced neurotoxicity in developing brain. However, the mechanisms are not extremely clear. To gain a better insight into the toxic effects of propofol on hippocampal neurons, we treated cells at the days in vitro 7 (DIV 7), which were prepared from Sprague-Dawley embryos at the 18th day of gestation, with propofol (0.1-1000M) for 3h. A significant decrease in neuronal proliferation and a remarkable increase in neuroapoptosis were observed in DIV 7 hippocampal neurons as measured by 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide assay and apoptosis assay respectively. Moreover, propofol treatment decreased the nuclear factor kappaB (NF-B) p65 expression, which was accompanied by a reduction in B-cell lymphoma 2 (Bcl-2) mRNA and protein levels, increased caspase-3 mRNA and activation of caspase-3 protein. These results indicated that downregulation of NF-B p65 and Bcl-2 were involved in the potential mechanisms of propofol-induced neurotoxicity. This likely led to the caspase-3 activation, triggered apoptosis and inhibited the neuronal growth and proliferation that we have observed in our in vitro systems. Copyright (c) 2014 John Wiley & Sons, Ltd.
BERNSM,SEEBERGL,SCHMIDTM,et al.High-dose propofol triggers short-term neuroprotection and long-term neurodegeneration in primary neuronal cultures from rat embryos[J].J Int Med Res,2009,37(3):680-688.
This study investigated the effects of propofol on primary neuronal cultures from rat embryos. Primary cortical neuronal cultures were prepared from Wistar rat embryos (E18). The viability of cells exposed to 0.01, 0.1 or 1 mg/ml propofol for up to 48 h was assessed using a methyltetrazolium assay. In order to evaluate the role of gamma-aminobutyric acid-A (GABA(A)) receptors, cells were also preincubated with the GABA(A)-receptor antagonists, gabazine and picrotoxin. Propofol at a concentration of 1 mg/ml significantly reduced cell viability after 12 h. In contrast, this concentration led to a significant increase in cell viability at 3 and 6 h. The GABA(A)-receptor antagonists did not influence the neurodegenerative effect of propofol but abolished its neuroprotective effect. DNA fragmentation as a marker of apoptosis was elevated after 24 h propofol treatment. These results confirm that high doses of propofol can cause GABA(A) receptor triggered neuroprotection and a subsequent time-dependent, but GABA(A) independent, neurodegeneration in primary cortical neurons.
YUEN VW.Dexmedetomidine:Perioperative application in children[J].Paediatr Anaesth,2010,20(3):256-264.
Dexmedetomidine is a highly specific and selective alpha-2-adrenergic agonist with sedative, anxiolytic, and organ protective effects. Its clinical applications in children include premedication, prevention of emergence delirium, as part of multimodal anesthetic regimen and sedation in the pediatric intensive care unit. Its role in neuroprotection in children undergoing anesthesia should be explored. In this review, various uses of dexmedetomidine are discussed in detail.
Abstract BACKGROUND: Anesthetic agents (eg, isoflurane, propofol) may cause neurodegeneration in the developing brains and impair animals' learning ability. Dexmedetomidine (DEX), a selective alpha 2-adrenoreceptor agonist, has antiapoptotic properties in several brain injury models. Here, we tested whether DEX can protect the brain from neurodegeneration in rats exposed to propofol in utero. MATERIALS AND METHODS: Fetal rats of embryonic day 20 were exposed in utero for 1 hour to propofol anesthesia with DEX or saline, or no anesthesia (control). The fetal brains were harvested 6 hours later. Cleaved caspase-3 levels and the relative number of ionized calcium-binding adaptor molecule 1 (IBA1)-positive cells were assessed by Western blot and immunohistochemistry. Learning and memory functions of the offspring in a separate cohort were assessed at postnatal day 35 by using an 8-arm radial maze. RESULTS: Propofol anesthesia in pregnant rats augmented caspase-3 activation by 217% in the brain tissues of fetal rats and increased the number of IBA1-positive cells in the cortex by 40% and in the thalamus by 270%. Juvenile rats exposed prenatally to propofol were not different than controls on spontaneous locomotor activity, but made more errors of omission and took longer to complete visiting all 8 arms on days 1, 2, and 3 across a 5-day test in the radial arm maze. This neurocognitive deficit was prevented by administration of DEX (5.0 碌g/kg, IP), which also significantly inhibited propofol-induced caspase-3 activation and microglial response in the fetal brains. CONCLUSIONS: DEX attenuates neuronal injury induced by maternal propofol anesthesia in the fetal brains, providing neurocognitive protection in the offspring rats.
LIUF,RAINOSEK SW,SADOVOVAN,et al.Protective effect of acetyl-L-carnitine on propofol-induced toxicity in embryonic neural stem cells[J].Neurotoxi,2014,42:49-57.
Propofol is a widely used general anesthetic. A growing body of data suggests that perinatal exposure to general anesthetics can result in long-term deleterious effects on brain function. In the developing brain there is evidence that general anesthetics can cause cell death, synaptic remodeling, and altered brain cell morphology. Acetyl- l -carnitine ( l -Ca), an anti-oxidant dietary supplement, has been reported to prevent neuronal damage from a variety of causes. To evaluate the ability of l -Ca to protect against propofol-induced neuronal toxicity, neural stem cells were isolated from gestational day 14 rat fetuses and on the eighth day in culture were exposed for 2402h to propofol at 10, 50, 100, 300 and 60002μM, with or without l -Ca (1002μM). Markers of cellular proliferation, mitochondrial health, cell death/damage and oxidative damage were monitored to determine: (1) the effects of propofol on neural stem cell proliferation; (2) the nature of propofol-induced neurotoxicity; (3) the degree of protection afforded by l -Ca; and (4) to provide information regarding possible mechanisms underlying protection. After propofol exposure at a clinically relevant concentration (5002μM), the number of dividing cells was significantly decreased, oxidative DNA damage was increased and a significant dose-dependent reduction in mitochondrial function/health was observed. No significant effect on lactase dehydrogenase (LDH) release was observed at propofol concentrations up to 10002μM. The oxidative damage at 5002μM propofol was blocked by l -Ca. Thus, clinically relevant concentrations of propofol induce dose-dependent adverse effects on rat embryonic neural stem cells by slowing or stopping cell division/proliferation and causing cellular damage. Elevated levels of 8-oxoguanine suggest enhanced oxidative damage [reactive oxygen species (ROS) generation] and l -Ca effectively blocks at least some of the toxicity of propofol, presumably by scavenging oxidative species and/or reducing their production.
STRAIKO MM,YOUNGC,CATTANOD,et al.Lithium protects against anesthesia-induced developmental neuroapoptosis[J].Anesthesiology,2009,110(4):862-868.
BACKGROUND: Ethanol and anesthetic drugs trigger neuroapoptosis in the developing mouse brain. Recently, it was found that ethanol-induced neuroapoptosis is preceded by suppressed phosphorylation of extracellular signal-regulated protein kinase (ERK), and lithium counteracts both the phosphorylated ERK suppressant action and ethanol-induced neuroapoptosis. The current study was undertaken to address the following questions. (1) Do ketamine and propofol mimic ethanol in suppressing ERK phosphorylation? (2) If they do, does lithium prevent this suppressant action and also prevent these anesthetic drugs from triggering neuroapoptosis? METHOD: Postnatal day 5 mice were treated with propofol, ketamine, lithium, a combination of propofol or ketamine with lithium or saline, and their brains were prepared for Western blot analysis or histology. For Western blot, cytosolic lysates of caudate putamen were analyzed for expression of phosphorylated ERK and phosphorylated serine/threonine-specific protein kinase. For histology, brains were stained immunohistochemically with antibodies to activated caspase-3, and the density of activated caspase-3 positive cells was determined. RESULTS: Ketamine and propofol suppressed phosphorylated ERK, and lithium counteracted both the phosphorylated ERK suppressant action and neuroapoptotic action of these anesthetic drugs. CONCLUSION: If further testing finds lithium to be safe for use in pediatric/obstetric medicine, administration of a single dose of lithium before anesthesia induction may be a suitable means of mitigating the risk of anesthesia-induced developmental neuroapoptosis.
SIFRINGERM,VON HAEFENC,KRAINM,et al.Neuro-protective effect of dexmedetomidine on hyperoxia-induced toxicity in the neonatal rat brain[J].Oxid Med Cell Longev,2015:530371.
Abstract Dexmedetomidine is a highly selective agonist of α2-receptors with sedative, anxiolytic, analgesic, and anesthetic properties. Neuroprotective effects of dexmedetomidine have been reported in various brain injury models. In the present study, we investigated the effects of dexmedetomidine on neurodegeneration, oxidative stress markers, and inflammation following the induction of hyperoxia in neonatal rats. Six-day-old Wistar rats received different concentrations of dexmedetomidine (1, 5, or 106508g/kg bodyweight) and were exposed to 80% oxygen for 2465h. Sex-matched littermates kept in room air and injected with normal saline or dexmedetomidine served as controls. Dexmedetomidine pretreatment significantly reduced hyperoxia-induced neurodegeneration in different brain regions of the neonatal rat. In addition, dexmedetomidine restored the reduced/oxidized glutathione ratio and attenuated the levels of malondialdehyde, a marker of lipid peroxidation, after exposure to high oxygen concentration. Moreover, administration of dexmedetomidine induced downregulation of IL-1β on mRNA and protein level in the developing rat brain. Dexmedetomidine provides protections against toxic oxygen induced neonatal brain injury which is likely associated with oxidative stress signaling and inflammatory cytokines. Our results suggest that dexmedetomidine may have a therapeutic potential since oxygen administration to neonates is sometimes inevitable.
DUANX,LIY,ZHOUC,et al.Dexmedetomidine provides neuroprotection: impact on ketamine-induced neuroa-poptosis in the developing rat brain[J].Acta Anaesthesiol Scand,2014,58(9):1121-1126.
Ketamine and dexmedetomidine are increasingly used in combination in pediatric patients. This study examined the hypothesis that dexmedetomidine attenuated ketamine‐induced neurotoxicity.
LIY,ZENGM,CHENW,et al.Dexmedetomidine reduces isoflurane-induced neuroapoptosis partly by preserving PI3K/Akt pathway in the hippocampus of neonatal rats[J].PLoS One,2014,9(4):e93639.
Prolonged exposure to volatile anesthetics, such as isoflurane and sevoflurane, causes neurodegeneration in the developing animal brains. Recent studies showed that dexmedetomidine, a selective 伪2-adrenergic agonist, reduced isoflurane-induced cognitive impairment and neuroapoptosis. However, the mechanisms for the effect are not completely clear. Thus, we investigated whether exposure to isoflurane or sevoflurane at an equivalent dose for anesthesia during brain development causes different degrees of neuroapoptosis and whether this neuroapoptosis is reduced by dexmedetomidine via effects on PI3K/Akt pathway that can regulate cell survival. Seven-day-old (P7) neonatal Sprague-Dawley rats were randomly exposed to 0.75% isoflurane, 1.2% sevoflurane or air for 6 h. Activated caspase-3 was detected by immunohistochemistry and Western blotting. Phospho-Akt, phospho-Bad, Akt, Bad and Bcl-xL proteins were detected by Western blotting in the hippocampus at the end of exposure. Also, P7 rats were pretreated with various concentrations of dexmedetomidine alone or together with PI3K inhibitor LY294002, and then exposed to 0.75% isoflurane. Terminal deoxyribonucleotide transferase-mediated dUTP nick end labeling (TUNEL) and activated caspase-3 were used to detect neuronal apoptosis in their hippocampus. Isoflurane, not sevoflurane at the equivalent dose, induced significant neuroapoptosis, decreased the levels of phospho-Akt and phospho-Bad proteins, increased the expression of Bad protein and reduced the ratio of Bcl-xL/Bad in the hippocampus. Dexmedetomidine pretreatment dose-dependently inhibited isoflurane-induced neuroapoptosis and restored protein expression of phospho-Akt and Bad as well as the Bcl-xL/Bad ratio induced by isoflurane. Pretreatment with single dose of 75 碌g/kg dexmedetomidine provided a protective effect similar to that with three doses of 25 碌g/kg dexmedetomidine. Moreover, LY294002, partly inhibited neuroprotection of dexmedetomidine. Our results suggest that dexmedetomidine pretreatment provides neuroprotection against isoflurane-induced neuroapoptosis in the hippocampus of neonatal rats by preserving PI3K/Akt pathway activity.
KOOE,OSHODIT,MESCHTERC,et al.Neurotoxic effects of dexmedetomidine in fetal cynomolgus monkey brain[J].J Toxicol Sci,2014,39(2):251-262.
The neuroprotective effects of dexmedetomidine have been reported by many investigators; however its underlying mechanism to reduce neuronal injury during a prolonged anesthesia remains unclear. In this study, we investigated the neurotoxic effects of dexmedetomidine in fetal monkey brains. In the present study, we compare the neurotoxic effects of dexmedetomidine and ketamine, a general anesthetic with a different mechanism of action, in fetal cynomolgus monkeys. Twenty pregnant monkeys at approximate gestation day 120 were divided into 4 groups: non-treatment controls (Group 1); ketamine at 20 mg/kg intramuscularly followed by a 12-hr infusion at 20-50 mg/kg/hr (Group 2); dexmedetomidine at 3 mu g/kg intravenously (i.v.) over 10 min followed by a 12-hr infusion at the human equivalent dose (HED) of 3 mu g/kg/hr (Group 3); and dexmedetomidine at 30 mu g/kg i.v. over 10 min followed by a 12-hr infusion at 30 mu g/kg/hr, 10 times HED (Group 4). Blood samples from both dams and fetuses were measured for concentration of dexmedetomidine. Each fetus was perfusion-fixed, serial sections were cut through the frontal cortex, and stained to detect for apoptosis (activated caspase 3 and TUNEL) and neurodegeneration (silver stain). In utero treatment with ketamine resulted in marked apoptosis and degeneration primarily in layers I and II of the frontal cortex. In contrast, fetal brains from animals treated with dexmedetomidine showed none to minimal neuroapoptotic or neurodegenerative lesions at both low- and high-dose treatments. Plasma levels confirmed systemic exposure of dexmedetomidine in both dams and fetuses. In conclusion, these results demonstrate that dexmedetomidine at both low-dose (RED) and high-dose (10 times HED) does not induce apoptosis in the frontal cortex (layers I, II, and III) of developing brain of cynomolgus monkeys.
ZOUW,XIAOH,GUM,et al.Propofol induces rat embr-yonic neural stem cell apoptosis by activating both extrinsic and intrinsic pathways[J].Mol Med Rep,2013,7(4):1123-1128.
Propofol has previously been shown to have detrimental effects on the developing brain. Neural stem cells, identified in the embryonic brain as well as in the adult brain, are multipotent, self-renewing cells, which are capable of differentiating into different phenotypes of the nervous system. The present study was designed to investigate propofol-induced rat embryonic neural stem cell apoptosis and its potential mechanisms. Rat embryonic neural stem cells were isolated, cultured and characterized. Treatment of these cultured stem cells with different doses of propofol was carried out and cell proliferation was assessed by MTT assay and apoptosis by flow cytometric analysis. Cellular levels of active forms of caspase-3 and caspase-8, which regulate the extrinsic apoptotic pathway, and of caspase-9 and cytochrome C, which regulate the intrinsic apoptotic pathway, were detected by western blotting. Over 95% of isolated rat embryonic neural stem cells expressed the Nestin protein, as detected by immunofluorescence staining. Using an in vitro cell culture system, we showed that propofol inhibited cell growth and induced cell apoptosis in a dose-dependent manner. Furthermore, western blot analysis showed that propofol treatment significantly elevated levels of active forms of caspase-3, caspase-8, caspase-9 and cytochrome C in the embryonic neural stem cells. Propofol induced rat embryonic neural stem cell apoptosis and activated caspase-3, caspase-8, caspase-9 and cytochrome C, suggesting that propofol-induced stem cell apoptosis may be regulated through both the extrinsic and intrinsic apoptotic pathways.
HAEBERLEIN SL.Mitochondrial function in apoptotic neu-ronal cell death[J].Neurochem Res,2004,29(3):521-530.
<a name="Abs1"></a>Apoptosis can be defined as the regulated death of a cell and is conducted by conserved pathways. Apoptosis of neurons after injury or disease differs from programed cell death, in the sense that neurons in an adult brain are not <img src="/content/JL706053756X3163/xxlarge8220.gif" alt="ldquo" align="MIDDLE" border="0">meant<img src="/content/JL706053756X3163/xxlarge8221.gif" alt="rdquo" align="MIDDLE" border="0"> to die and results in a loss of function. Thus apoptosis is an honorable process by a neuron, a cell with limited potential to replace itself, choosing instead to commit suicide to save neighboring cells from release of cellular components that cause injury directly or trigger secondary injury resulting from inflammatory reactions. The excess of apoptosis of neuronal cells underlies the progressive loss of neuronal populations in neurodegenerative disorders and thus is harmful. Mitochondria are the primary source for energy in neurons but are also poised, through the <img src="/content/JL706053756X3163/xxlarge8220.gif" alt="ldquo" align="MIDDLE" border="0">mitochondrial apoptosis pathway,<img src="/content/JL706053756X3163/xxlarge8221.gif" alt="rdquo" align="MIDDLE" border="0"> to signal the demise of cells. This duplicity of mitochondria is discussed, with particular attention given to the specialized case of pathological neuronal cell death.
BARACCAA,SGARBIG,SOLAINIG,et al.Rhodamine 123 as a probe of mitochondrial membrane potential: evaluation of proton flux through F(0) during ATP synthesis[J].Biochim Biophys Acta,2003,1606(1-3):137-146.
Abstract Rhodamine 123 (RH-123) was used to monitor the membrane potential of mitochondria isolated from rat liver. Mitochondrial energization induces quenching of RH-123 fluorescence and the rate of fluorescence decay is proportional to the mitochondrial membrane potential. Exploiting the kinetics of RH-123 fluorescence quenching in the presence of succinate and ADP, when protons are both pumped out of the matrix driven by the respiratory chain complexes and allowed to diffuse back into the matrix through ATP synthase during ATP synthesis, we could obtain an overall quenching rate proportional to the steady-state membrane potential under state 3 condition. We measured the kinetics of fluorescence quenching by adding succinate and ADP in the absence and presence of oligomycin, which abolishes the ADP-driven potential decrease due to the back-flow of protons through the ATP synthase channel, F(0). As expected, the initial rate of quenching was significantly increased in the presence of oligomycin, and conversely preincubation with subsaturating concentrations of the uncoupler carbonyl cyanide p-trifluoro-metoxyphenilhydrazone (FCCP) induced a decreased rate of quenching. N,N'-dicyclohexylcarbodiimide (DCCD) behaved similarly to oligomycin in increasing the rate of quenching. These findings indicate that RH-123 fluorescence quenching kinetics give reliable and sensitive evaluation of mitochondrial membrane potential, complementing steady-state fluorescence measurements, and provide a mean to study proton flow from the mitochondrial intermembrane space to the matrix through the F(0) channel.
... 丙泊酚通过激动γ氨基丁酸A型受体(Gamma amino acid type A receptor,GABAR)和抑制N-甲基-D-天冬氨酸受体(N-methyl-D-aspartic acid receptor,NMDAR)发挥麻醉作用,《中华人民共和国药典》2010年版提示丙泊酚慎用于<3岁患儿,但该药具有起效快、苏醒快、并发症少等优点,在实际临床工作中仍被广泛应用于婴幼儿麻醉的诱导与维持.近年来大量动物实验和细胞研究表明,丙泊酚对发育期大脑具有神经毒性[1-4],因此丙泊酚临床应用,尤其是在婴幼儿麻醉中的应用,引起广泛关注.本研究发现,丙泊酚可对原代培养皮质神经元产生损伤,表现为神经元形态学损伤,细胞存活率下降,细胞凋亡率上升,出现细胞核固缩和裂解等细胞凋亡的典型表现. ...
Effect of propofol in the immature rat brain on short-and long-term neurodevelopmental outcome
2013
Propofol inhibits proliferation and induces neuroapoptosis of hippocampal neurons in vitro via downregulation of NF-κB p65 and Bcl-2 and upregulation of caspase-3
2014
High-dose propofol triggers short-term neuroprotection and long-term neurodegeneration in primary neuronal cultures from rat embryos
... 丙泊酚通过激动γ氨基丁酸A型受体(Gamma amino acid type A receptor,GABAR)和抑制N-甲基-D-天冬氨酸受体(N-methyl-D-aspartic acid receptor,NMDAR)发挥麻醉作用,《中华人民共和国药典》2010年版提示丙泊酚慎用于<3岁患儿,但该药具有起效快、苏醒快、并发症少等优点,在实际临床工作中仍被广泛应用于婴幼儿麻醉的诱导与维持.近年来大量动物实验和细胞研究表明,丙泊酚对发育期大脑具有神经毒性[1-4],因此丙泊酚临床应用,尤其是在婴幼儿麻醉中的应用,引起广泛关注.本研究发现,丙泊酚可对原代培养皮质神经元产生损伤,表现为神经元形态学损伤,细胞存活率下降,细胞凋亡率上升,出现细胞核固缩和裂解等细胞凋亡的典型表现. ...
Dexmedetomidine:Perioperative application in children
... 针对麻醉药引起的发育期大脑损伤,美国国立卫生研究院(national institutes of health,NIH)、美国食品药品管理局(food and drug administration,FDA) 以及国际麻醉研究学会(international anesthesia research society,IARS)要求不仅要研究麻醉药神经毒性的发生机制,而且要寻找有效的措施防治麻醉药引起的发育期神经损伤.众多学者对如何防治丙泊酚对婴幼儿大脑产生的神经损伤进行了广泛研究,发现锂剂、乙酰左旋肉碱等对丙泊酚引起的神经元损伤有保护作用[7-8],但它们在婴幼儿使用的安全性有待进一步探讨.盐酸右美托咪定是另一高选择性α2肾上腺素受体激动药,具有镇静镇痛作用,作为麻醉辅助药物广泛应用于临床麻醉.目前大量研究发现,盐酸右美托咪定具有神经保护作用,如对缺血-再灌注损伤[9],高氧诱导的发育期大脑损伤[10]产生保护作用.另有动物实验研究表明,盐酸右美托咪定可对麻醉药氯胺酮、异氟烷以及丙泊酚引起的发育期大鼠大脑凋亡样损伤产生保护作用,但盐酸右美托咪定减轻麻醉药发育期神经毒性的机制目前还不清楚[6,11-12] .另有研究认为,盐酸右美托咪定本身对发育期大脑不产生神经损伤作用[13].盐酸右美托咪定是否对抗丙泊酚诱导的原代培养皮质神经元损伤,笔者未见报道,故在本实验中对此进行了研究. ...
Protective effect of acetyl-L-carnitine on propofol-induced toxicity in embryonic neural stem cells
1
2014
... 针对麻醉药引起的发育期大脑损伤,美国国立卫生研究院(national institutes of health,NIH)、美国食品药品管理局(food and drug administration,FDA) 以及国际麻醉研究学会(international anesthesia research society,IARS)要求不仅要研究麻醉药神经毒性的发生机制,而且要寻找有效的措施防治麻醉药引起的发育期神经损伤.众多学者对如何防治丙泊酚对婴幼儿大脑产生的神经损伤进行了广泛研究,发现锂剂、乙酰左旋肉碱等对丙泊酚引起的神经元损伤有保护作用[7-8],但它们在婴幼儿使用的安全性有待进一步探讨.盐酸右美托咪定是另一高选择性α2肾上腺素受体激动药,具有镇静镇痛作用,作为麻醉辅助药物广泛应用于临床麻醉.目前大量研究发现,盐酸右美托咪定具有神经保护作用,如对缺血-再灌注损伤[9],高氧诱导的发育期大脑损伤[10]产生保护作用.另有动物实验研究表明,盐酸右美托咪定可对麻醉药氯胺酮、异氟烷以及丙泊酚引起的发育期大鼠大脑凋亡样损伤产生保护作用,但盐酸右美托咪定减轻麻醉药发育期神经毒性的机制目前还不清楚[6,11-12] .另有研究认为,盐酸右美托咪定本身对发育期大脑不产生神经损伤作用[13].盐酸右美托咪定是否对抗丙泊酚诱导的原代培养皮质神经元损伤,笔者未见报道,故在本实验中对此进行了研究. ...
Lithium protects against anesthesia-induced developmental neuroapoptosis
1
2009
... 针对麻醉药引起的发育期大脑损伤,美国国立卫生研究院(national institutes of health,NIH)、美国食品药品管理局(food and drug administration,FDA) 以及国际麻醉研究学会(international anesthesia research society,IARS)要求不仅要研究麻醉药神经毒性的发生机制,而且要寻找有效的措施防治麻醉药引起的发育期神经损伤.众多学者对如何防治丙泊酚对婴幼儿大脑产生的神经损伤进行了广泛研究,发现锂剂、乙酰左旋肉碱等对丙泊酚引起的神经元损伤有保护作用[7-8],但它们在婴幼儿使用的安全性有待进一步探讨.盐酸右美托咪定是另一高选择性α2肾上腺素受体激动药,具有镇静镇痛作用,作为麻醉辅助药物广泛应用于临床麻醉.目前大量研究发现,盐酸右美托咪定具有神经保护作用,如对缺血-再灌注损伤[9],高氧诱导的发育期大脑损伤[10]产生保护作用.另有动物实验研究表明,盐酸右美托咪定可对麻醉药氯胺酮、异氟烷以及丙泊酚引起的发育期大鼠大脑凋亡样损伤产生保护作用,但盐酸右美托咪定减轻麻醉药发育期神经毒性的机制目前还不清楚[6,11-12] .另有研究认为,盐酸右美托咪定本身对发育期大脑不产生神经损伤作用[13].盐酸右美托咪定是否对抗丙泊酚诱导的原代培养皮质神经元损伤,笔者未见报道,故在本实验中对此进行了研究. ...
右美托咪定减轻大鼠全脑缺血再灌注损伤
1
2013
... 针对麻醉药引起的发育期大脑损伤,美国国立卫生研究院(national institutes of health,NIH)、美国食品药品管理局(food and drug administration,FDA) 以及国际麻醉研究学会(international anesthesia research society,IARS)要求不仅要研究麻醉药神经毒性的发生机制,而且要寻找有效的措施防治麻醉药引起的发育期神经损伤.众多学者对如何防治丙泊酚对婴幼儿大脑产生的神经损伤进行了广泛研究,发现锂剂、乙酰左旋肉碱等对丙泊酚引起的神经元损伤有保护作用[7-8],但它们在婴幼儿使用的安全性有待进一步探讨.盐酸右美托咪定是另一高选择性α2肾上腺素受体激动药,具有镇静镇痛作用,作为麻醉辅助药物广泛应用于临床麻醉.目前大量研究发现,盐酸右美托咪定具有神经保护作用,如对缺血-再灌注损伤[9],高氧诱导的发育期大脑损伤[10]产生保护作用.另有动物实验研究表明,盐酸右美托咪定可对麻醉药氯胺酮、异氟烷以及丙泊酚引起的发育期大鼠大脑凋亡样损伤产生保护作用,但盐酸右美托咪定减轻麻醉药发育期神经毒性的机制目前还不清楚[6,11-12] .另有研究认为,盐酸右美托咪定本身对发育期大脑不产生神经损伤作用[13].盐酸右美托咪定是否对抗丙泊酚诱导的原代培养皮质神经元损伤,笔者未见报道,故在本实验中对此进行了研究. ...
Neuro-protective effect of dexmedetomidine on hyperoxia-induced toxicity in the neonatal rat brain
1
2015
... 针对麻醉药引起的发育期大脑损伤,美国国立卫生研究院(national institutes of health,NIH)、美国食品药品管理局(food and drug administration,FDA) 以及国际麻醉研究学会(international anesthesia research society,IARS)要求不仅要研究麻醉药神经毒性的发生机制,而且要寻找有效的措施防治麻醉药引起的发育期神经损伤.众多学者对如何防治丙泊酚对婴幼儿大脑产生的神经损伤进行了广泛研究,发现锂剂、乙酰左旋肉碱等对丙泊酚引起的神经元损伤有保护作用[7-8],但它们在婴幼儿使用的安全性有待进一步探讨.盐酸右美托咪定是另一高选择性α2肾上腺素受体激动药,具有镇静镇痛作用,作为麻醉辅助药物广泛应用于临床麻醉.目前大量研究发现,盐酸右美托咪定具有神经保护作用,如对缺血-再灌注损伤[9],高氧诱导的发育期大脑损伤[10]产生保护作用.另有动物实验研究表明,盐酸右美托咪定可对麻醉药氯胺酮、异氟烷以及丙泊酚引起的发育期大鼠大脑凋亡样损伤产生保护作用,但盐酸右美托咪定减轻麻醉药发育期神经毒性的机制目前还不清楚[6,11-12] .另有研究认为,盐酸右美托咪定本身对发育期大脑不产生神经损伤作用[13].盐酸右美托咪定是否对抗丙泊酚诱导的原代培养皮质神经元损伤,笔者未见报道,故在本实验中对此进行了研究. ...
Dexmedetomidine provides neuroprotection: impact on ketamine-induced neuroa-poptosis in the developing rat brain
1
2014
... 针对麻醉药引起的发育期大脑损伤,美国国立卫生研究院(national institutes of health,NIH)、美国食品药品管理局(food and drug administration,FDA) 以及国际麻醉研究学会(international anesthesia research society,IARS)要求不仅要研究麻醉药神经毒性的发生机制,而且要寻找有效的措施防治麻醉药引起的发育期神经损伤.众多学者对如何防治丙泊酚对婴幼儿大脑产生的神经损伤进行了广泛研究,发现锂剂、乙酰左旋肉碱等对丙泊酚引起的神经元损伤有保护作用[7-8],但它们在婴幼儿使用的安全性有待进一步探讨.盐酸右美托咪定是另一高选择性α2肾上腺素受体激动药,具有镇静镇痛作用,作为麻醉辅助药物广泛应用于临床麻醉.目前大量研究发现,盐酸右美托咪定具有神经保护作用,如对缺血-再灌注损伤[9],高氧诱导的发育期大脑损伤[10]产生保护作用.另有动物实验研究表明,盐酸右美托咪定可对麻醉药氯胺酮、异氟烷以及丙泊酚引起的发育期大鼠大脑凋亡样损伤产生保护作用,但盐酸右美托咪定减轻麻醉药发育期神经毒性的机制目前还不清楚[6,11-12] .另有研究认为,盐酸右美托咪定本身对发育期大脑不产生神经损伤作用[13].盐酸右美托咪定是否对抗丙泊酚诱导的原代培养皮质神经元损伤,笔者未见报道,故在本实验中对此进行了研究. ...
Dexmedetomidine reduces isoflurane-induced neuroapoptosis partly by preserving PI3K/Akt pathway in the hippocampus of neonatal rats
1
2014
... 针对麻醉药引起的发育期大脑损伤,美国国立卫生研究院(national institutes of health,NIH)、美国食品药品管理局(food and drug administration,FDA) 以及国际麻醉研究学会(international anesthesia research society,IARS)要求不仅要研究麻醉药神经毒性的发生机制,而且要寻找有效的措施防治麻醉药引起的发育期神经损伤.众多学者对如何防治丙泊酚对婴幼儿大脑产生的神经损伤进行了广泛研究,发现锂剂、乙酰左旋肉碱等对丙泊酚引起的神经元损伤有保护作用[7-8],但它们在婴幼儿使用的安全性有待进一步探讨.盐酸右美托咪定是另一高选择性α2肾上腺素受体激动药,具有镇静镇痛作用,作为麻醉辅助药物广泛应用于临床麻醉.目前大量研究发现,盐酸右美托咪定具有神经保护作用,如对缺血-再灌注损伤[9],高氧诱导的发育期大脑损伤[10]产生保护作用.另有动物实验研究表明,盐酸右美托咪定可对麻醉药氯胺酮、异氟烷以及丙泊酚引起的发育期大鼠大脑凋亡样损伤产生保护作用,但盐酸右美托咪定减轻麻醉药发育期神经毒性的机制目前还不清楚[6,11-12] .另有研究认为,盐酸右美托咪定本身对发育期大脑不产生神经损伤作用[13].盐酸右美托咪定是否对抗丙泊酚诱导的原代培养皮质神经元损伤,笔者未见报道,故在本实验中对此进行了研究. ...
Neurotoxic effects of dexmedetomidine in fetal cynomolgus monkey brain
1
2014
... 针对麻醉药引起的发育期大脑损伤,美国国立卫生研究院(national institutes of health,NIH)、美国食品药品管理局(food and drug administration,FDA) 以及国际麻醉研究学会(international anesthesia research society,IARS)要求不仅要研究麻醉药神经毒性的发生机制,而且要寻找有效的措施防治麻醉药引起的发育期神经损伤.众多学者对如何防治丙泊酚对婴幼儿大脑产生的神经损伤进行了广泛研究,发现锂剂、乙酰左旋肉碱等对丙泊酚引起的神经元损伤有保护作用[7-8],但它们在婴幼儿使用的安全性有待进一步探讨.盐酸右美托咪定是另一高选择性α2肾上腺素受体激动药,具有镇静镇痛作用,作为麻醉辅助药物广泛应用于临床麻醉.目前大量研究发现,盐酸右美托咪定具有神经保护作用,如对缺血-再灌注损伤[9],高氧诱导的发育期大脑损伤[10]产生保护作用.另有动物实验研究表明,盐酸右美托咪定可对麻醉药氯胺酮、异氟烷以及丙泊酚引起的发育期大鼠大脑凋亡样损伤产生保护作用,但盐酸右美托咪定减轻麻醉药发育期神经毒性的机制目前还不清楚[6,11-12] .另有研究认为,盐酸右美托咪定本身对发育期大脑不产生神经损伤作用[13].盐酸右美托咪定是否对抗丙泊酚诱导的原代培养皮质神经元损伤,笔者未见报道,故在本实验中对此进行了研究. ...
Propofol induces rat embr-yonic neural stem cell apoptosis by activating both extrinsic and intrinsic pathways

 , 郭洪霞
, 郭洪霞
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}