中国科技论文统计源期刊 中文核心期刊
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖

 , 王雪萍, 崔锋, 郝海军
, WANG Xueping, CUI Feng,, HAO Haijun
, 王雪萍, 崔锋, 郝海军
, WANG Xueping, CUI Feng,, HAO Haijun
目的 建立快速、准确的测定盐酸厄洛替尼含量的氢核磁共振定量分析方法。方法 以马来酸为内标,DMSO-
Objective To establish a rapid and accurate method for quantitative determination of erlotinib hydrochloride by proton nuclear magnetic resonance (1H-NMR). Methods Maleic acid was used as an internal standard,and DMSO-
核磁共振(nuclear magnetic resonance, NMR)技术应用于定量分析的原理是化学环境不同的氢质子共振峰面积只与其氢原子个数相关,无需引入任何校正因子,于1963年开始用于定量分析[1]。随着高强度磁场和傅里叶变换技术的应用,使得仪器的性能得到极大改善,从而进一步推动了NMR技术应用于定量分析领域。目前,中国、英国及美国等各国药典均已收载了氢核磁共振定量法(quantitative nuclear magnetic resonance, qNMR)。盐酸厄洛替尼(Erlotinib hydrochloride)化学名为N-(3-乙炔基苯基)-6,7-二(2-甲氧基乙氧基)-4-喹唑啉胺盐酸盐,是一种分子靶向治疗药物,通过抑制人体细胞内表皮生长因子上的一种特定酶活性从而达到抗肿瘤作用[2-4]。其含量测定主要采用高效液相色谱法(HPLC)[5-6],但国内目前尚无官方提供的盐酸厄洛替尼对照品。在合成盐酸厄洛替尼时难免会引入其他结构相似的杂质,使用质量平衡法测定盐酸厄洛替尼含量时,需要HPLC法对杂质进行有效分离,引入校正因子才能得出HPLC法含量,同时还需测定其水分、残留溶剂及炽灼残渣等。操作过程繁琐,耗时费力;而qNMR法测定样品含量时无需对照品,不受样品中水分、残留溶剂等干扰,操作过程简单,结构准确,因此与HPLC法相比具有较大的优势[5]。本研究采用qNMR法对盐酸厄洛替尼样品含量进行了系统的方法学验证,为盐酸厄洛替尼原料药质量标准完善提供有力佐证,也为其对照品含量标定提供参考。
Bruker-400ADVENCE II型核磁共振仪(瑞士布鲁克公司);Topspin3.2试验控制及数据处理软件;高效液相色谱仪(安捷伦科技有限公司,型号:Agilent 1260);卡氏水分仪(瑞士万通中国有限公司,型号:915 KF Ti-touch);分析天平(德国Sartorius公司,感量:0.1 mg)。
氘代DMSO(DMSO-
精密称取马来酸对照品55.25 mg,溶于25 mL DMSO-
采用zg30脉冲序列测试1H-NMR。实验参数设置为:温度为300 K,谱宽(SWH)8012 Hz,点数(TD)64 K,采样时间(AT)4.0 s,脉冲宽度9.54 μs,弛豫延迟时间(D1)20 s,采样次数(NS)64次。
色谱柱:Waters C18(250 mm×4.6 mm,5 μm);流动相:甲醇-pH3.5磷酸盐缓冲液(20:80,v/v);流速1.0 mL·min-1;检测波长343 nm;柱温30 ℃;进样量10 μL[6]。
2.4.1 盐酸厄洛替尼1H-NMR谱图的归属 盐酸厄洛替尼在CDCl3,CD3OD等溶剂中溶解度很差,但溶于DMSO-
2.4.2 专属性实验 选用马来酸作为内标,与盐酸厄洛替尼混合物1H-NMR谱图见
2.4.3 线性关系考察 精密称取盐酸厄洛替尼样品2.88,4.95,7.78,14.24,19.08 mg,分别加入马来酸内标液1.0 mL溶解,混匀后,转移0.5 mL置5 mm核磁管中。以盐酸厄洛替尼的 δ8.84(B2)和δ 8.47(B1)定量峰的平均值与马来酸δ 6.26内标峰面积比(
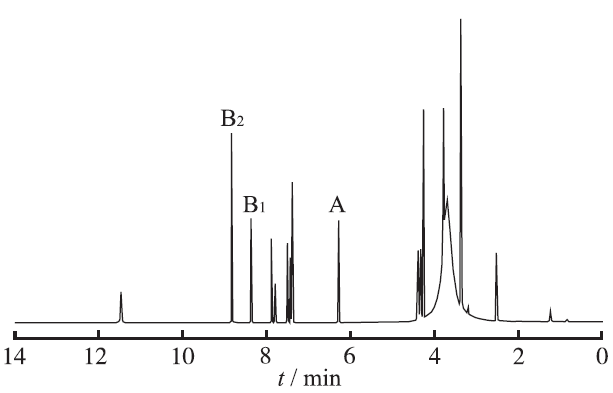
图2
马来酸与盐酸厄洛替尼1H-NMR谱图
A.内标特征峰;B1和B2.盐酸厄洛替尼特征峰
Fig.2
1H-NMR spectrum of erlotinib hydrochloride and erlotinib maleic acid
A.characteristic peak of internal standard;B1 and B2.characteristic peak of erlotinib hydrochloride
2.4.4 精密度实验 取“2.1”项下供试品溶液,按“2.2”项下条件连续进样测定6次,获取盐酸厄洛替尼与马来酸混合物的1H-NMR图谱,积分5次取其平均值,计算定量峰与内标峰面积的比值。计算RSD为0.36%。
2.4.5 重复性实验 按“2.1”项下方法平行制备6份供试品溶液。按照“2.2”项下条件进样测定,获取盐酸厄洛替尼与马来酸混合物1H-NMR图谱,积分5次取其平均值,计算定量峰与内标峰面积的比值。计算RSD为0.83%。
2.4.6 样品稳定性实验 取“2.1”项下供试品溶液,分别于0,2,4,8,12 h进行测定,获取盐酸厄洛替尼与马来酸混合物1H-NMR,积分5次取其平均值,计算定量峰与内标峰面积的比值。计算RSD为1.17%。
2.4.7 加样回收率实验 取6份已知含量的盐酸厄洛替尼样品,分别加入相同量的盐酸厄洛替尼工作对照品(含量:97.43%),并加入内标溶液配制样品溶液,按照“2.2”项下的测试条件获取1H-NMR,计算回收率。结果平均回收率为100.92%,RSD为1.74%。见
表1 盐酸厄洛替尼样品加样回收率实验
Tab.1 Recovery test of erlotinib hydrochloride
精密称取3份盐酸厄洛替尼,按“2.1”项下方法制备供试品溶液,按“2.2”项下测试条件进行测定,获取盐酸厄洛替尼与马来酸混合物1H-NMR,以盐酸厄洛替尼δ8.84(B2)和δ 8.47(B1)处峰信号的平均值和δ 6.26处马来酸峰信号,按照如下公式计算盐酸厄洛替尼含量:
含量(%)=
式中
经计算,3份盐酸厄洛替尼含量分别为92.26%,91.94%和92.09%,平均含量为92.10%,RSD为0.17%。
采用HPLC面积归一化法测定盐酸厄洛替尼色谱纯度为99.46%,干燥失重法测定样品含水量为7.25%,炽灼残渣结果为0.07%。根据质量平衡法计算,盐酸厄洛替尼(%)=(100%-水分%-炽灼残渣%)×HPLC纯度=92.17%[8]。
在确定弛豫时间,采样次数等重要的仪器参数时,尽管
采用qNMR定量也有一定的限制:待测化合物的结构必须已知才能正确定量;无法区分结构非常接近的类似物,定量峰必须与其他信号完全分离,才能准确定量;不适用于浓度极低的样品,这与qNMR灵敏度不高有关。但随着核磁共振技术的进一步发展,必然会推动qNMR技术被越来越多的学者所青睐,迎来更为广阔的应用前景。
The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}