中国科技论文统计源期刊 中文核心期刊
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖

 , 郑成, ZHENG Cheng
, 郑成, ZHENG Cheng目的 建立小儿热速清颗粒的质量标准。方法 采用薄层色谱法(TLC)对小儿热速清颗粒中的大黄、柴胡、连翘、葛根进行定性鉴别;采用高效液相色谱(HPLC)法测定小儿热速清颗粒中黄芩苷的含量。结果 建立了专属性良好的大黄、柴胡、连翘、葛根的薄层鉴别方法。黄芩苷在0.116 2~1.743 0 μg范围内呈良好的线性关系,
Objective To establish the quality standard of
小儿热速清颗粒由柴胡、黄芩、金银花、板蓝根、葛根、大黄、连翘、水牛角8味中药组成,具有清热解毒、泻火利咽功效,主要用于小儿外感风热所致感冒,症见高热、头痛、咽喉肿痛、鼻塞流涕、咳嗽、大便干结等。小儿热速清颗粒为2013年国家基本药物目录品种,现行标准收载于《国家药品标准新药转正第38册》,标准编号为WS3-092(Z-092)-2002(Z),包括理化鉴别、黄芩和大黄的薄层鉴别及黄芩苷的含量测定[1]。由于现行标准未对处方中君药柴胡和葛根、连翘等其他药味进行控制,且原标准中大黄的薄层鉴别由于前处理中未进行酸水解,色谱斑点不显著,黄芩苷含量测定的色谱条件有待改进,故笔者根据国家标准提高研究课题任务书要求,修订了大黄的薄层鉴别方法,新增了柴胡、葛根和连翘的薄层鉴别方法,修订了黄芩苷含量测定方法,以更好地控制该制剂的质量。
日本岛津LC-20AT高效液相色谱仪,二极管阵列检测器(DAD);Mettler240型电子分析天平。
黄芩苷对照品(批号:110715-201117,纯度:91.7%)、葛根对照药材(批号:1175-200001)、连翘对照药材(批号:120908-201216)、柴胡对照药材(批号:0992-200102)、大黄对照药材(批号:1249-0301)、黄芩对照药材(批号:120955-200406)、大黄素对照品(批号:0756-200110)、葛根素对照品(批号:110752-200912)、连翘苷对照品(批号:0821-9903),均由中国食品药品检定研究院提供;甲醇为色谱纯;水为重蒸水,其他试剂均为分析纯。薄层色谱用硅胶G板、硅胶GF254板均由青岛海洋化工厂提供,小儿热速清颗粒15批样品分别由哈尔滨圣泰药业有限公司和江西倍肯药业有限公司提供。(批号:2013042701,2013052503,2013052701,2013052702,2013052902,2013053002,2013053102,2016053103,2013060101,2013060103,20130624,20130701,20130702,20130703,20130704)。
取本品5 g,研细,加甲醇40 mL,超声处理30 min,滤过,滤液蒸干,残渣加水20 mL使溶解,加盐酸2 mL,加热回流30 min,立即冷却,用乙醚振摇提取2次,每次20 mL,合并乙醚液,蒸干,残渣加甲醇2 mL使溶解,作为供试品溶液。另取大黄对照药材0.1 g,同法制成对照药材溶液。再取大黄素对照品,加甲醇制成每毫升含0.5 mg的溶液,作为对照品溶液。根据处方取缺大黄的其他各味药材,按法制成颗粒,取颗粒适量,照供试品溶液的制备方法制成阴性对照溶液。照薄层色谱法实验,分别吸取供试品溶液、阴性对照溶液各10 μL及对照药材溶液、对照品溶液各1 μL,分别点于同一硅胶H薄层板上,以石油醚(30~60 ℃)-甲酸乙酯-甲酸(15∶5∶1)的上层溶液为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视[2]。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点,阴性对照无干扰,结果见
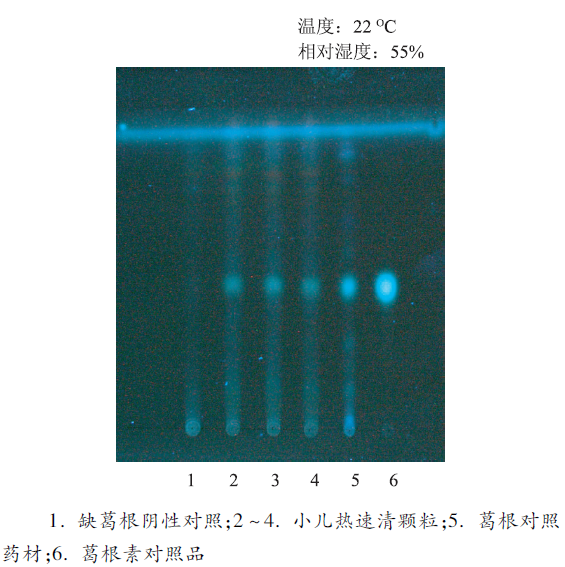
取本品5 g,研细,加乙酸乙酯50 mL,超声处理30 min,滤过,滤液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。取葛根对照药材0.5 g,同供试品溶液制备方法制备对照药材溶液。另取葛根素对照品,加甲醇制成每毫升含0.5 mg的溶液,作为对照品溶液。根据处方取缺葛根的其他各味药材,按法制成颗粒,取颗粒适量,照供试品溶液制备方法制成阴性对照溶液。照薄层色谱法实验,吸取供试品溶液、阴性对照溶液各10 μL及对照品溶液5 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(7∶2.5∶0.25)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视[3]。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点,阴性对照无干扰,结果见
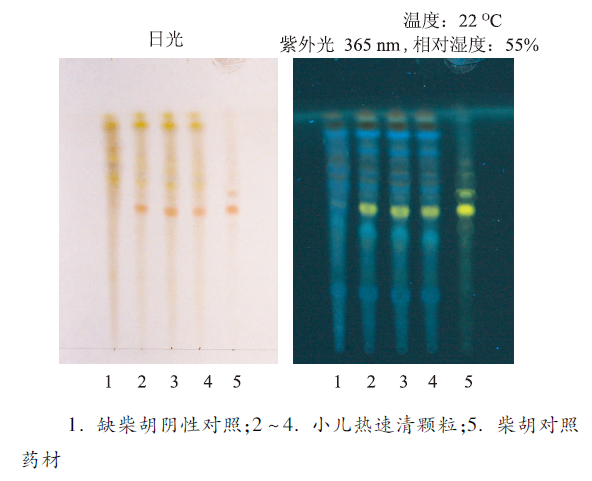
取本品5 g,研细,加水40 mL使溶解,用水饱和的正丁醇振摇提取3次,每次30 mL,合并正丁醇提取液,用氨试液洗涤2次,每次30 mL,弃去氨试液,正丁醇液蒸干,残渣加甲醇2 mL使溶解,静置,取上清液,作为供试品溶液。另取柴胡对照药材0.5 g,加水40 mL,加热微沸1 h,滤过,从“用水饱和的正丁醇振摇提取3次”起,同法制成对照药材溶液。根据处方取缺柴胡的其他各味药材,按法制成颗粒,取颗粒适量,照供试品溶液的制备方法制成阴性对照溶液。照薄层色谱法实验,分别吸取供试品溶液、阴性对照溶液各10 μL及对照药材溶液5 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(13∶7∶2)10 ℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以2%对二甲氨基苯甲醛的40%硫酸溶液,在60 ℃加热至斑点显色清晰,分别置日光及紫外光灯(365 nm)下检视[4]。供试品色谱中,在与对照药材色谱相应的位置上,日光下显相同颜色的斑点;紫外光灯下显相同颜色的荧光斑点,阴性对照无干扰,结果见
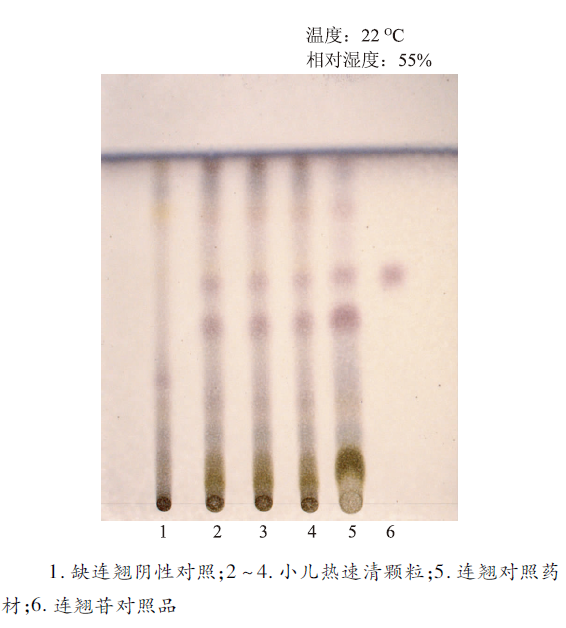
取本品5 g,研细,加水30 mL使溶解,离心10 min(3 000 r·min-1),通过D101型大孔吸附树脂柱(柱内径为2 cm,柱高为15 cm),以1.5 mL·min-1流速,先后用水150 mL和甲醇100 mL洗脱,收集甲醇洗脱液,蒸干,残渣加甲醇2 mL使溶解,静置,取上清液,作为供试品溶液。另取连翘对照药材1 g,加甲醇10 mL,加热回流30 min,滤过,滤液浓缩至约2 mL,作为对照药材溶液。再取连翘苷对照品,加甲醇制成每毫升含1 mg的溶液,作为对照品溶液。根据处方取缺连翘的其他各味药材,按法制成颗粒,取颗粒适量,照供试品溶液的制备方法制成阴性对照溶液。照薄层色谱法实验,吸取供试品溶液、阴性对照溶液各10 μL及对照药材溶液、对照品溶液各5 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-醋酸 (16∶3∶1)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105 ℃加热至斑点显色清晰[5]。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点,阴性对照无干扰,结果见
色谱柱:Agilent-Extend C18(250 mm×4.6 mm,5 μm);流动相:甲醇-水(48∶52)为流动相;检测波长:277 nm;流速:1.0 mL·min-1;柱温:30 ℃;进样量:10 μL。理论板数按黄芩苷峰计算,应不低于2 500。
取黄芩苷对照品适量,精密称定,置200 mL量瓶中,加50%甲醇适量,置热水浴中振摇使溶解,放冷,加50%甲醇至刻度,摇匀,制得每毫升含50 μg的溶液,即得。
取本品,研细,取约0.5 g,精密称定,置100 mL量瓶,精密加入50%甲醇40 mL,分别超声处理(功率250 W,25 kHz)15 min,放冷,用50%甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
根据处方取缺黄芩的其他各味药材,按法制成颗粒,取颗粒适量,照供试品溶液的制备方法制成阴性对照溶液。
3.5.1 专属性考察 按“3.1”项色谱条件测定黄芩苷对照品溶液、供试品溶液和缺黄芩阴性对照溶液,阴性对照溶液色谱图中未出现与黄芩苷保留时间一致的色谱峰,表明方法专属性良好,见
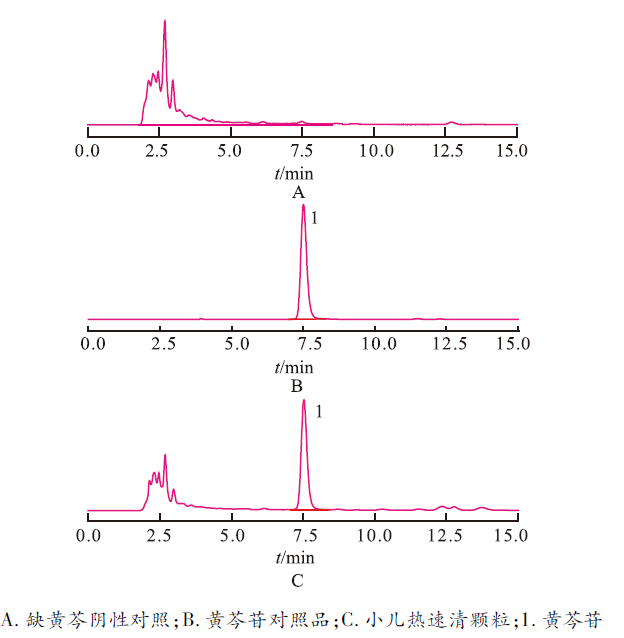
图5
专属性实验结果
A.negative reference without
Fig.5 Results of specificity test
3.5.2 线性关系考察 精密吸取116.20 μg·mL-1黄芩苷对照品溶液1,2,4,8,10,15 μL,按“3.1”项方法进样测定峰面积。以色谱峰面积为纵坐标,以进样量(μg)为横坐标,进行线性回归,得线性方程为:
3.5.3 精密度实验 精密吸取供试品溶液,连续进样6次,测定峰面积,计算得RSD为0.15%。结果表明,该方法精密度良好。
3.5.4 重复性实验 取同一批样品(批号:2013042701),精密称定6份,按“3.3”项方法制备供试品溶液,测定。结果黄芩苷平均含量为每袋32.62 mg(RSD=0.14%,
3.5.5 稳定性实验 精密吸取供试品溶液,分别在0,2,4,8,12,18,24 h进样测定,计算。结果黄芩苷含量的RSD为0.53%,表明样品溶液至少24 h内稳定。
3.5.6 加样回收率实验 取同一批样品(批号:2013042701)约0.25 g,平行6份,精密称定,分别精密加入122.8 μg·mL-1黄芩苷对照品溶液40 mL,按“3.3”项方法制备供试品溶液,进样测定,计算回收率。结果平均回收率100.61%,RSD为0.79%,见
表1 小儿热速清颗粒中黄芩苷加样回收率实验结果
Tab.1
Result of recovery test
按“3.3”项下方法制备15批样品的供试品溶液并进样测定,结果见
表2 小儿热速清颗粒中黄芩苷含量测定结果
Tab.2
Results of content determination on baicalin in
在确定薄层色谱鉴别方法时,笔者从样品制备、展开剂组成及比例的选择、点样量、不同品牌薄层板、展开环境(如温度和湿度)等方面,对几味药材的薄层色谱条件进行了优化。结果显示,所采用的为最佳实验条件。
笔者分别比较了50%甲醇、30%乙醇、50%乙醇、75%乙醇4种提取溶剂及超声15和30 min对含量的影响。结果表明,不同提取溶剂和超声时间对含量结果影响较小,考虑到流动相组成及提取效率,选择50%甲醇作为提取溶剂,超声提取15 min。
笔者比较了3种不同品牌色谱柱及柱温25,30,35和40 ℃时对样品测定的影响。结果供试品溶液在Diamonsil-C18(250 mm×4.6 mm,5 μm)、Agilent Extend-C18(250 mm×4.6 mm,5 μm)和Welch-C18(250 mm×4.6 mm,5 μm)色谱柱上均能得到良好分离,且不同柱温仅对出峰时间有影响,表明所建立的含量测定方法对色谱柱耐用性良好。
从15批样品的测定结果看,两家企业黄芩苷含量接近,但不同规格含量差异较大,故优化黄芩苷含量测定方法并制定合理限度对保证产品质量有意义。
The authors have declared that no competing interests exist.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}