中国科技论文统计源期刊 中文核心期刊
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖

 , 张宇佳, 陈谕园, 张楠, 郑稳生
, ZHANG Yujia, CHEN Yuyuan, ZHANG Nan, ZHENG Wensheng
, 张宇佳, 陈谕园, 张楠, 郑稳生
, ZHANG Yujia, CHEN Yuyuan, ZHANG Nan, ZHENG Wensheng
目的 建立测定左旋多巴聚乙二醇(PEG)化固体脂质纳米粒包封率的方法,进行质量评价。方法 采用葡聚糖凝胶(Sephadex G-50)微柱离心法分离固体脂质纳米粒和游离药物;高效液相色谱(HPLC)法测定固体脂质纳米粒中左旋多巴含量,计算包封率。结果 在所建立的色谱条件下,辅料不干扰测定,左旋多巴在10.54~527.00 μg·mL-1范围内线性关系良好,高、中、低浓度加样回收率分别为99.13%,99.51%,99.04%,RSD分别为1.25%,1.91%,1.71%;所建立的微柱离心条件能有效分离左旋多巴固体脂质纳米粒与游离药物。空白固体脂质纳米粒的洗脱率为98.84%,RSD 为0.80%(
Objective To establish a mini-column centrifugation-HPLC method to determine the entrapment efficiency of levodopa-loaded PEGylated-solid lipid nanoparticles. Methods A dextran gel(Sephadex G-50) mini-column centrifugation was employed to separate the free drug from solid lipid nanoparticles.The content of levodopa was qualified by HPLC. Results Under the applied chromatographic condition,the excipients had no influence on the determination of levodopa.A calibrated linear of levodopa concentration was within 10.54-527.00 μg·mL-1.The recoveries of high,medium and low concentrations of levodopa were 99.13%,99.51% and 99.04%(RSD were 1.25%,1.91% and 1.71%), respectively.The free levodopa was well separated from solid lipid nanoparticles by using mini-column centrifugation.The addition of blank solid lipid nanoparticles recovery was 98.84% with RSD of 0.80%(
目前临床主要使用的抗帕金森病药物为左旋多巴(levodopa,L-DOPA),其本身并无药理活性,透过血-脑屏障进入中枢后经多巴脱羧酶分解转化为多巴胺而发挥药理作用。然而左旋多巴口服生物利用度和脑摄取率很低,主要是因为在外周循环中被芳香族氨基酸大量水解,临床服用左旋多巴片剂后约2 h血药浓度即降至有效浓度(0.6~24 μg·mL-1)以下[1-2],常与外周脱羧酶抑制药联合给药。固体脂质纳米粒(solid lipid nanoparticles,SLN)以天然或合成的固态类脂化合物为载体,将药物包裹于类脂核中制成的固态胶粒系统,可以提高药物稳定性,此外还具有可控释放、靶向定位释放、毒性低等优点[3]。聚乙二醇(polyethylene glycol,PEG)是一种无毒、无免疫原性和抗原性的亲水性化合物,对药物的释放无影响。载体经低分子质量PEG修饰后,血浆半衰期延长。有研究显示,SLN表面用低分子质量高密度PEG修饰后,粒子疏水性和静电作用均降低。笔者以聚乙二醇化单硬脂酸为脂材,采用复乳-溶剂挥发法制备载左旋多巴的PEG化SLN(LDP-pSLN),以期提高LDP的口服生物利用度,延长其血浆半衰期同时获得平稳的血药浓度。为控制LDP-pSLN的质量,采用微柱离心-HPLC法测定其包封率,方法简捷,结果重复性好。
ZNCL-BS180磁力搅拌仪(北京瑞威伟业仪器设备有限公司);Hitachi高效液相色谱仪(四元泵、在线脱气机、自动进样器、DAD检测器,日本日立公司);低速离心机(湖南湘仪实验仪器开发有限公司);紫外分光光度计(北京普析通用仪器有限责任公司);380ZLS粒径仪(美国PSS公司)。
左旋多巴(上海阿拉丁试剂公司,批号:74847335);三硬脂酸甘油酯(上海安谱试剂公司,批号:X322G005);聚乙二醇单硬脂酸酯(上海阿拉丁试剂公司,批号:H1503075);胆固醇(国药集团化学试剂公司,批号:20141029);大豆卵磷脂(国药集团化学试剂公司,批号:20141204);Brij S10(上海阿拉丁试剂,批号:G1404054);乙腈为色谱纯,其他试剂均为分析纯。
采用复乳-溶剂挥发法制备LDP-pSLN。精密称取处方量三硬脂酸甘油酯、聚乙二醇单硬脂酸酯,75 ℃水浴融化作为油相,将此油相、内水相 LDP的盐酸溶液以及相同温度的大豆卵磷脂/正丁醇溶液混合,轻度搅拌形成W/O微乳[4];得到的微乳缓慢滴加于400 r·min-1搅拌下40 ℃的Brij S10水溶液中,形成W/O/W复乳,挥干有机溶剂后继续搅拌至体积浓缩为原来的四分之一时,将得到的半透明体系进行冰浴固化,2 h后以300 W的功率探头超声10 min,得LDP-pSLN悬液。
除以纯化水代替LDP盐酸溶液作为内水相外,按“2.1”项方法制备,即得Blank-pSLN。
Blank-pSLN破乳液:精密量取Blank-pSLN悬液0.1 mL,置于10 mL量瓶,加无水乙醇0.5 mL超声5 min破乳,1%盐酸定容至刻度,摇匀,即得。
LDP对照品溶液:精密称取LDP10 mg,置于10 mL量瓶,1%盐酸溶解并定容至刻度,摇匀,即得。
LDP-pSLN破乳液:精密量取LDP-pSLN悬液0.1 mL,置10 mL量瓶,加无水乙醇0.5 mL超声30 min破乳,1%盐酸定容至刻度,摇匀,即得。
2.4.1 色谱条件 色谱柱:Kromasil C18柱(4.6 mm×250 mm,5 μm),流动相:0.1%三氟乙酸溶液-乙腈(96:4),流速:1.0 mL·min-1,检测波长:280 nm,进样量:10 μL,柱温:25 ℃。
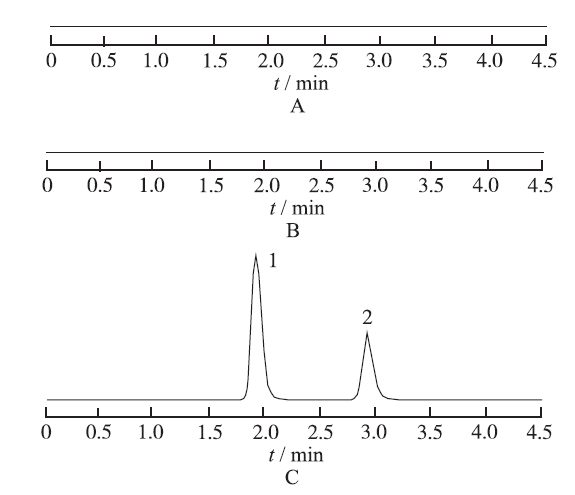
2.4.2 专属性考察 取LDP对照品溶液、Blank-pSLN破乳液以及LDP-pSLN破乳液各10 μL,按“2.4.1”项色谱条件进样测定,记录色谱图,见

图1
3种溶液HPLC图
A.空白PEG化固体脂质纳米粒;B.左旋多巴对照品溶液;C.左旋多巴PEG化固体脂质纳米粒;1.左旋多巴
Fig.1
HPLC chromatograms of three kinds of solution
A.blank PEG-solid lipid nanoparticle;B.levodopa control;C.levodopa PEG- solid lipid nanoparticle;1.levodopa
2.4.3 线性与范围 精密称取LDP10.54 mg,置10 mL量瓶,用1%盐酸溶解并定容至刻度,摇匀,作为贮备液,分别稀释制成10.54,21.08,42.16,52.70,105.4,210.8,527.0 μg·mL-1供试品溶液,按“2.4.1”项色谱条件进样测定,记录峰面积。以LDP浓度(
2.4.4 仪器精密度实验 精密量取52.70 μg·mL-1对照品溶液,重复进样测定6次,记录峰面积,计算得RSD为0.49%(
2.4.5 重复性实验 取同一批(批号:20160415)样品1 mL,平行6份,置于10 mL量瓶,加无水乙醇0.5 mL破乳后用1%盐酸定容至刻度,摇匀,按“2.4.1”项色谱条件进样测定。结果LDP平均含量46.01 μg·mL-1,RSD为0.62%(
2.4.6 稳定性实验 取同一批(批号:20160415)样品,按“2.4.5”项方法制备供试品溶液,在室温下放置0,1,2,4,6,8,10,12,24 h后分别进样检测,记录峰面积,计算RSD为0.57%(
2.4.7 方法回收率实验 分别精密量取“2.4.3”项储备液0.1,0.3,1.0 mL各3份,置10 mL量瓶,加入Blank-pSLN悬液0.2 mL,加无水乙醇0.5 mL破乳后用1%盐酸稀释并定容至刻度,摇匀,按“2.4.1”项色谱条件进样测定,记录峰面积,计算加样回收率。高、中、低浓度加样回收率分别为99.13%,99.51%,99.04%,RSD分别为1.25%,1.91%,1.71%,表明该方法准确度良好。
2.5.1 微型凝胶柱的制备 称取一定量Sephadex G-50用纯化水溶胀12 h后,装入2 mL去掉内塞的注射器中(底部放两张略小于其内径的圆形滤纸)排除气泡后,以水平衡2~3个柱体积[5],以3 000 r·min-1(
2.5.2 微型凝胶柱对Blank-pSLN的吸附 取Blank-pSLN 0.1 mL 缓缓加入到微柱的顶端,500 r·min-1离心1 min(
表1 Blank-pSLN的微柱离心回收率
Tab.1
Recovery of Blank-pSLN by mini-column after centrifugation
2.5.3 微柱对游离药物的吸附 精密称取 L-DOPA原料药3份,以1%盐酸溶解并定容,配制成浓度分别为0.1,0.5,1 mg·mL-1溶液。精密吸取各浓度溶液0.1 mL,缓缓加入微柱顶端,按“2.5.2”项条件离心洗脱,合并收集的洗脱液置于10 mL量瓶,用1%盐酸定容至刻度,摇匀,按“2.4.1”项色谱条件分别进样测定,记录峰面积为
2.5.4 微柱对Blank-pSLN和游离药物物理混合液的吸附 精密量取0.1 mL的空白SLN 3份置于10 mL 量瓶,分别加入适量L-DOPA原料药,用1%盐酸定容至刻度,制成浓度分别为0.1,0.5,1 mg·mL-1物理混合液。按“2.4.1”项色谱条件测定过柱前药物的峰面积(
2.5.5 洗脱曲线的绘制 精密吸取LDP-pSLN悬液0.1 mL加到微柱顶端“2.4.2”项离心条件进行洗脱,收集每次的洗脱液,加无水乙醇破乳后用1%盐酸定容至10 mL,按“2.4.1”项色谱条件测定峰面积,根据峰面积与收集管数绘制洗脱曲线,其中1~4 管洗脱液为空白脂质体混悬液,5~14 管洗脱液为游离L-DOPA溶液,SephadexG-50 微型凝胶柱可较好地将SLN与游离L-DOPA药物分离,结果见
精密量取3 份不同批次的LDP-pSLN悬液各0.1 mL,上样于微柱顶端,按“2.5.2”项方法分离游离药物和SLN,最终洗脱液收集到10 mL 量瓶,经无水乙醇破乳后,用1%盐酸稀释定容至刻度,摇匀,按“2.4.1”项色谱条件测定包封于SLN中药物浓度
文献[6]报道,离心转速和时间以及微柱的高度对葡聚糖凝胶微柱的分离效果有很大的影响。转速过高或离心的时间过长会使凝胶溶胀吸收的水分离出来,导致凝胶柱中出现大量气泡甚至断裂;转速过低或离心时间过短会减少洗脱体积,导致S纳米粒和游离药物分离效果不好。微柱的高度过高导致洗脱体积增大,稀释倍数增大,给破乳带来不便;微柱的高度过低,分离效果不好,易导致游离药物洗出。本实验对3种规格的葡聚糖凝胶(SephadexG-25,50 ,100)进行了考察,并对离心力、离心时间和柱高进行反复摸索,筛选出微柱离心分离的最佳条件为微柱高度1.7 cm,3 000 r·min-1离心3 min。
紫外分光光度法对Blank-pSLN的破乳液进行全波长扫描,显示其在208 nm波长处有最大吸收,故选择208 nm测定Blank-pSLN破乳液的吸光度。
The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}