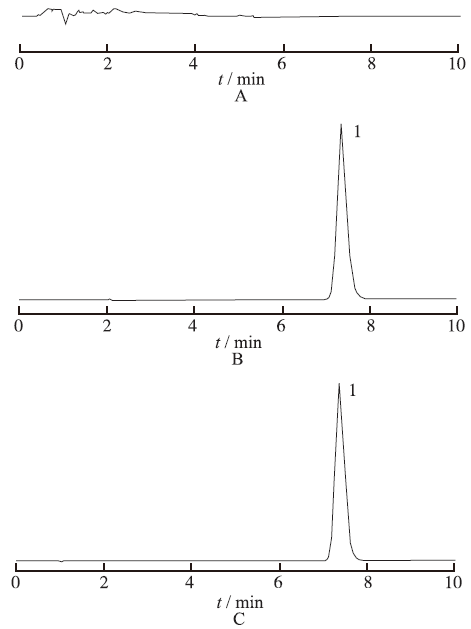
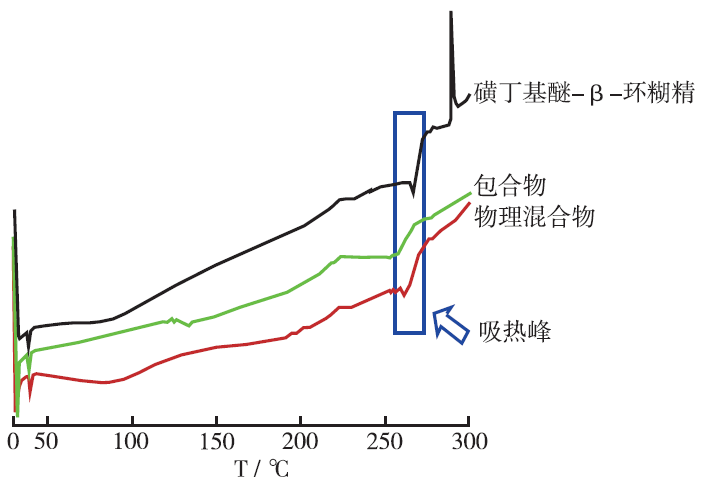
Objective To optimize preparation process of butylphthalide sulfobutyl ether-β-cyclodextrin inclusion complex. Methods Freeze-drying method was adopted to prepare butylphthalide sulfobutyl ether-β-cyclodextrin inclusion complex. Inclusion ratio was served as index, the effects of inclusion temperature, inclusion time and the ratio of sulfobutyl ether-β-cyclodextrin to butylphthalide on the inclusion process were investigated by using response surface method. The optimal inclusion process was predicted according to the formulation which was established and fitted by multi-linear equation, second-order polynomial equation and the third-order polynomial equation. Results The optimum inclusion technology of butylphthalide sulfobutyl ether-β-cyclodextrin inclusion complex was as follows: inclusion temperature was 67 ℃, inclusion time was 2.09 h, the input ratio of sulfobutyl ether-β-cyclodextrin to butylphthalide was 2.6:1. The deviation of inclusion ratio between actual value and predicted value was 2.4%. Conclusion Mathematical model established by central composite design and response surface method has a good prediction, which can be used to optimize the preparation process of butylphthalide sulfobutyl ether-β-cyclodextrin inclusion complex.
DEVASARIN,DORA CP,SINGHC,et al.Inclusion complex of erlotinib with sulfobutyl ether-β-cyclodextrin:preparation,characterization,in silico,in vitro and in vivo evaluation[J].Carbohydr Polym,2015,134:547-556.
The aim of the study was to investigate the impact of erlotinib sulfobutyl ether beta-cyclodextrin complex (ERL-SBE-β-CD) on ERL dissolution rate and oral bioavailability. Preliminary comparative phase solubility study indicated ERL exhibited maximum solubility in SBE-β-CD solution. Optimal experimental design confirmed freeze drying of SBE-β-CD:ERL in 1:1.05 molar ratio as the optimum method. Differential scanning calorimetry (DSC), Fourier transformation infrared spectroscopy (FT-IR), powder X-ray diffractometry (PXRD), proton nuclear magnetic resonance (1H NMR) and two-dimensional rotating-frame Overhauser effect spectroscopy (2D ROESY NMR) confirmed the inclusion complexation. The in silico computational study, employed to analyze the comparative interactions of ERL with SBE-β-CD and β-CD, indicated ease of ERL-SBE-β-CD complexation. In vitro dissolution and in vivo bioavailability studies further confirmed the ERL-SBE-β-CD as a valuable approach to enhance ERL oral bioavailability with 3.6-fold increase in relative oral bioavailability with higher Cmax (134.29±36.51 vs. 42.36±1.75μg/ml) and AUC0–∞ (2103.47±156.75 vs.580.43±71.91μg/mlh) over the free drug. The complex exhibited 3.2-fold increase in Cmax with 5.4-fold decrease in Tmax (0.5±0.2 vs. 2.7±0.8h) in comparison to pure ERL. Thus, ERL-SBE-β-CD complexation exhibits a potential to enhance oral bioavailability of ERL leading to reduce dose and dose-related side effects.
XUC,TANGY,HUW,et al.Investigation of inclusion complex of honokiol with sulfobutyl ether-β-cyclodextrin[J].Carbohydr Polym,2014,113:9-15.
This study aimed to prepare and characterize an inclusion complex of honokiol (HNK) with sulfobutyl ether-β-cyclodextrin (SB-β-CD). The inclusion complex (HNK/CD COMP) was prepared utilizing a freeze–drying method. Phase-solubility curves were employed to obtain stability constants and thermodynamic parameters. The phase-solubility diagram showed a typical AL-type, indicating that the 1:1 (HNK:SB-β-CD) inclusion complex was formed. The solid inclusion complex was then characterized by differential scanning calorimetry and Fourier transform infrared spectroscopy. Results showed that HNK/CD COMP exhibited a higher drug release rate than free HNK in vitro. A comparative study of the pharmacokinetics between HNK/CD COMP and free HNK was also performed in rats. In vivo results indicated that AUC0–t and Cmax of HNK/CD COMP increased by approximately 158% and 123% compared with those of the free HNK, respectively. These results suggest that SB-β-CD will be potentially useful in the delivery of poorly soluble drugs, such as HNK.
RENL,ZHOUY,WEIP,et al.Preparation and pharma-cokinetic study of aprepitant-sulfobutyl ether-β-cyclodextrin complex[J].AAPS Pharm Sci Tech,2014,15(1):121-130.
Aprepitant (APR), a neurokinin 1 receptor antagonist, is an approved treatment for chemotherapy-induced nausea and vomiting and for post-operative nausea and vomiting. However, it has poor water solubility. This study was performed to optimize the capsule formulation of an inclusion complex of APR with sulfobutyl ether-β-cyclodextrin (SBE-β-CD), and to evaluate its water solubility, dissolution rate, and bioavailability. The complex was prepared through the saturated-aqueous solution method and then characterized by Fourier transform infrared spectroscopy, x-ray powder diffraction, and differential scanning calorimetry. Subsequently, a pharmacokinetic study was performed using liquid chromatography–tandem mass spectrometry. Emend, which features an innovative formulation that incorporates drug nanoparticles with high bioavailability, was used as a reference for comparison with the optimized formulation. As a result, the dissolution rates and extent of release of the test formulation in various media were enhanced relative to those of Emend. The bioavailability of the drug complex was comparable to that of Emend. In summary, the SBE-β-CD complexation could provide a practical and cost-effective option for enhancing the solubility and bioavailability of APR according to our research.
Atherosclerosis is an inflammatory disease linked to elevated blood cholesterol concentrations. Despite ongoing advances in the prevention and treatment of atherosclerosis, cardiovascular disease remains the leading cause of death worldwide. Continuous retention of apolipoprotein B–containing lipoproteins in the subendothelial space causes a local overabundance of free cholesterol. Because cholesterol accumulation and deposition of cholesterol crystals (CCs) trigger a complex inflammatory response, we tested the efficacy of the cyclic oligosaccharide 2-hydroxypropyl-β-cyclodextrin (CD), a compound that increases cholesterol solubility in preventing and reversing atherosclerosis. We showed that CD treatment of murine atherosclerosis reduced atherosclerotic plaque size and CC load and promoted plaque regression even with a continued cholesterol-rich diet. Mechanistically, CD increased oxysterol production in both macrophages and human atherosclerotic plaques and promoted liver X receptor (LXR)–mediated transcriptional reprogramming to improve cholesterol efflux and exert anti-inflammatory effects. In vivo, this CD-mediated LXR agonism was required for the antiatherosclerotic and anti-inflammatory effects of CD as well as for augmented reverse cholesterol transport. Because CD treatment in humans is safe and CD beneficially affects key mechanisms of atherogenesis, it may therefore be used clinically to prevent or treat human atherosclerosis.

 , 田伟
, 田伟
{kind=link}
{kind=link}
{kind=link}
{kind=link}