中国科技论文统计源期刊 中文核心期刊
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖

 , 刘欢, 杨悦
, LIU Huan, YANG Yue,
, 刘欢, 杨悦
, LIU Huan, YANG Yue,
采用文献分析法,从制度实施对象、实施方式和实施结果的角度介绍并分析日本新药再审查制度。日本厚生劳动省在批准新药时针对不同类型的新有效成分、剂量、给药途径或适应证药品指定不同的再审查期,再审查期内上市许可持有人须履行开展上市后调查等义务,再审查期结束后的3个月内提交再审查申请,厚生劳动省重新评估该药品的安全性和有效性。提炼出新药再审查制度每个环节的特点,提出完善我国新药上市后监测体系的启示。建议扩展新药监测范围;丰富再注册申请资料的检查方式;解决新药监测期后与药品再注册的关联问题。
To provide enlightenments for monitoring post-marketing new drugs in China. Through literature analysis, new drug re-examination system in Japan was introduced and analyzed from the perspective of system implementation objects, implementation methods and implementation results. The Japanese Ministry of Health, Labor and Welfare assigns specific re-examination periods for different types of new active ingredients, dosages, routes of administration and indications when approving new drugs. During the re-examination period, marketing authorization holders must fulfill their obligations to conduct post-marketing investigations. The application for re-examination is submitted within 3 months after the end of the re-examination period, and the Ministry of Health, Labour and Welfare re-evaluates the safety and effectiveness of the new drugs. After elaborating the characteristics of each link of the new drug re-examination periods, it puts forward some enlightenments perfecting the new drug monitoring system in China. It is recommended to expand the monitoring scope of new drugs, developing more inspection methods of re-registration application materials, and solve the problem of association with drug re-registration after the new drug monitoring period.
为收集、分析并评价已上市药品信息,掌握药品已知效益和风险,收集新的疗效、适应证和不良反应信息,解决药品上市前研究的局限性和上市后使用的复杂性问题,为公众提供安全、有效的药品,日本于1967年建立全国药物监测体系,并于1979年以法律形式确立了药品上市后监测制度(Post-marketing Surveil-ance,PMS),是亚洲第一个以法律形式确定药品上市后监测制度的国家[1]。日本PMS制度由新药再审查制度、药品再评价制度和药品不良反应(ADR)报告制度组成,笔者着重分析日本新药再审查制度。
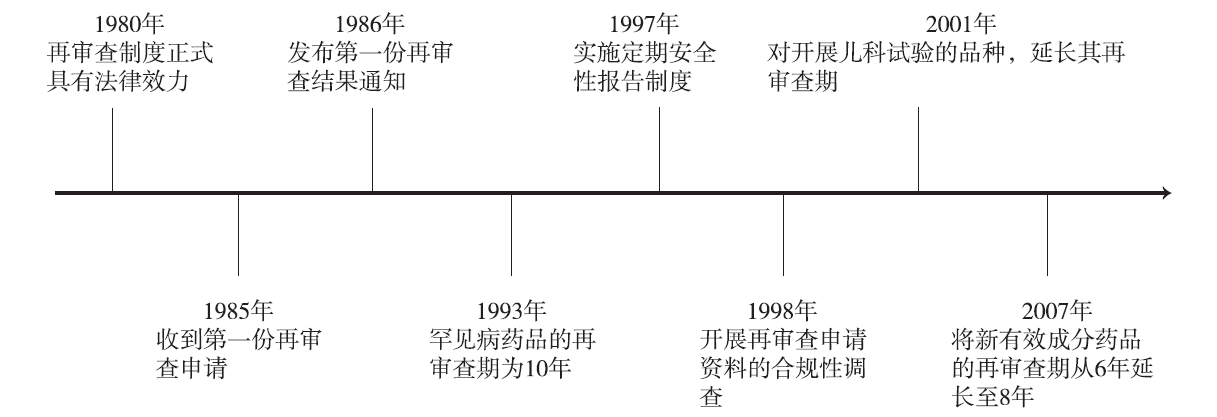
由于新药上市前的临床试验受试者数量有限,在临床试验中难以发现全部效益和风险,且新药临床试验中研究者能够对受试者的症状、年龄、用量和联合用药等进行选择和控制,但实际临床用药情况复杂,为此,日本建立了新药再审查制度。日本新药再审查制度可追溯到1967年厚生劳动省发布的《关于医药品制造许可标准基本规定》(厚生劳动省医药局文件第645号),该规定要求已获批新药的生产企业在该药品上市后的2年内收集其上市后不良反应等信息,2年期限结束后对该药品的安全性有效性进行重新审查。经过一系列的修订和完善,1980年4月,再审查制度正式建立,并写入日本的《药事法》中。日本新药再审查制度发展历程见
再审查是指出于对已获批新药的安全性、有效性等进行重新确认的目的,对新药从获批之日起一段时间内的使用情况等所进行的调查[2]。在法规体系方面,《药事法》《药品上市后研究质量管理规范》(good post-marketing study practice,GPSP)以及《药物警戒质量管理规范》(good vigilance practice,GVP)等共同保障再审查制度的顺利实施,其中GPSP对药品上市后为采集数据和资料用于再审查和再评价而进行的调查和试验进行管理,以确保已上市药品的上市许可持有人(marketing authorization holder,MAH)为再审查或再评价所提交的数据真实可靠[3];而GVP则是对药品上市后的安全监管建立了一系列标准,涉及到药品安全监管信息的收集和分析,以及安全保障措施的制定和实施等。在组织体系方面,日本厚生劳动省统筹负责日本新药再审查制度的实施,其组成部门药品和食品安全局(Pharmaceutical and Food Safety Bureau,PFSB)负责制定再审查相关政策,作为咨询顾问机构的药政管理和食品卫生委员会(Pharmaceutical Affairs and Food Sanitation Council,PAFSC)为日本厚生劳动省开展再审查工作提供相关建议,而独立法人机构药品医疗器械管理局(Pharmaceutical and Medical Devices Agency,PMDA)则负责对接受再审查药品进行技术审查。
根据《药事法》第78条要求,接受厚生劳动省再审查的药品的MAH必须缴纳再审查费用。不同再审查环节的费用不同,如对于GPSP合规性调查,日本国内申请人需缴纳费用270.72万日元,而日本国外申请人需缴纳费用297.42万日元和相应差旅费[4]。
《药事法》第14.4.1条对再审查制度的设立对象及再审查期做出了规定:与已获批药品的有效成分、剂量、给药途径或适应证等明显不同的药品即新药,其MAH必须在该药品的再审查期内开展使用情况调查等,并在再审查期结束后的3个月内,提交再审查申请,接受厚生劳动省的再审查。不同类型新药的再审查期不同,具体如下[5]:①罕见病药品的再审查期为10年;②新有效成分药品的再审查期为8年;③新给药途径药品的再审查期为6年;④新适应证、新剂量药品的再审查期为4~6年。对于某些特殊药品,如含有儿科剂量的药品,厚生劳动省大臣可在听取PAFSC的意见后,适当延长其再审查期,最长不超过10年。
日本设立新药再审查期,一方面是加大对新药的监管力度,通过在再审查期内收集新药的上市后信息,及时发现药品安全性问题,并在再审查期结束后重新评估新药的安全性有效性,将无效药品、问题药品撤市;另一方面是授予新药绝对市场独占,根据《关于医药品制造许可标准有关规定》[6],在新药再审查期内,厚生劳动省不受理该药品任何仿制药的上市申请,即使该药品的仿制药企业利用自行研究取得的数据申请仿制药上市。
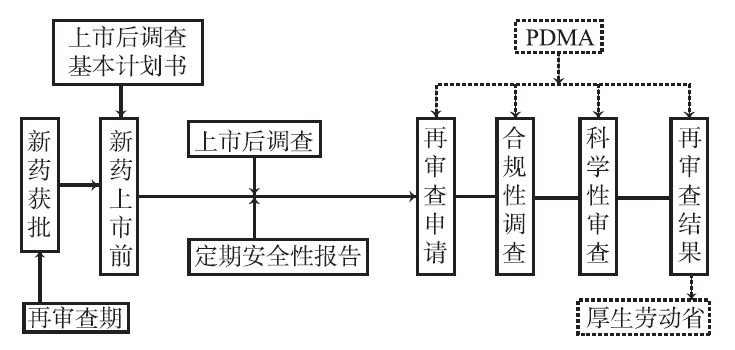
根据GPSP规定,MAH须在该药品拟定上市之日前至少1个月,向PMDA提交药品上市后调查基本计划书。基本计划书中包含MAH对该药品研发,海外类似药品安全性问题的分析,以及在再审查期内开展上市后调查的预方案。PMDA负责审核基本计划书,并确定最终的药品上市后调查方案[7]。
《药事法》第14.4.2.4条要求MAH根据厚生劳动省大臣制定的标准,在再审查期内调查收集新药上市后的使用情况等资料,并作为再审查申请资料的一部分提交给厚生劳动省。厚生劳动省大臣制定的标准即为GPSP。GPSP进一步明确了新药MAH须开展的上市后调查,将上市后调查定义为:MAH为收集、验证、确认与药品质量、安全性和有效性相关的信息所开展的药品使用情况调查或上市后临床试验。
3.2.1 使用情况调查 使用情况调查是一项基于日常诊疗的、无对照组的非干预性观察性调查。新药的MAH须与使用该药品的医疗机构签订调查合同,并向该医疗机构内开具该药品的医生寄发专门的使用情况调查表。调查内容包括用药患者的姓名、用药患者出现的已知或未知的ADR、在日常诊疗情况下患者使用该药品的有效性等。
MAH在进行使用情况调查时不应对用药患者设定条件,而应收集并分析所有用药患者出现的不良反应以及药品质量、有效性、安全性的相关信息,以发现该药品未知的不良反应,或已知不良反应发生频率的变化及其他安全性信息的信号等。
3.2.2 专项使用情况调查 专项使用情况调查是MAH针对儿童、老年人、孕妇、肾病或肝病患者、长期服药患者的用药信息的调查,调查内容和方式与使用情况调查相同,仅调查对象不同,以获取在研发过程中无法获取或难以获取的数据。
3.2.3 上市后临床试验 为验证通过日常诊疗、使用情况调查、专项使用情况调查以及其他用药信息所得到的与该药品相关的推断和假设,或获取在日常诊疗中无法获取的、合理的用药信息,厚生劳动省要求MAH开展新药上市后临床试验。新药上市后临床试验与为申请上市而开展的临床试验的要求一致,仅开展试验的时间点不同,MAH必须遵守GPSP以及《药物临床试验质量管理规范》(Good Clinical Practice,GCP),并在该药品已获批的用法用量、效果效能范围内开展新药上市后临床试验。
合规性调查由实地调查和文件调查两部分组成。《药事法》第14.4.2.5条规定根据厚生劳动大臣制定的标准(即GPSP)对MAH提交的再审查申请资料进行实地调查或文件调查,以确保再审查申请资料的可靠性。PMDA负责根据GPSP开展合规性调查,其中实地调查主要是对新药的MAH是否设置上市后调查责任人,是否制定操作程序手册(包括:①使用情况以及专项使用情况调查程序;②上市后临床试验程序;③自查程序;④上市后调查人员的教育培训程序;⑤上市后调查的外包程序;⑥上市后调查记录的保存程序;⑦其他能够确保上市后调查顺利进行的必要程序)等相关事项进行检查;而文件调查则是检查申请资料是否符合GPSP规定,并追溯原始资料和记录,如调查MAH与医疗机构签订的调查合同是否合理,调查表中的描述是否正确,调查结果的总结和分析是否正确等。PMDA在完成再审查申请资料的合规性调查后,发布合规性调查结果通知。
在接受合规性调查后,合格的再审查申请资料还需接受PMDA新药审评小组的科学性审查。新药审评小组针对申请资料中存在的科学性问题向MAH提出询问,并根据回复进行科学性审查,将科学性审查结果提交给厚生劳动省。
日本厚生劳动省最终收到的再审查申请资料应由以下几部分组成:由MAH提交的再审查申请资料概要;再审查申请的附加资料(包括使用情况调查资料、专项使用情况调查资料、上市后临床试验资料、ADR报告、文献研究报告、国内外对该药品所采取的措施报告以及严重药害事件报告)和参考资料,以及PMDA提交的合规性调查结果和科学性审查结果[8]。
根据《药事法》第74.2条,日本厚生劳动省有权对接受再审查的新药做出撤销上市批准,删除或修改药品部分获批事项或药品通过再审查的决定,并发布最终的再审查结果通知。当日本厚生劳动省认为删除或修改药品部分获批事项在卫生保健上有必要时,可做出删除或修改该药品部分获批事项的决定;新药的MAH必须在收到再审查结果通知的2周内,提交新药获批事项部分变更申请。当日本厚生劳动省认为接受再审查的药品已不符合《药事法》第14条要求时,即不具备已获批适应证时,或其风险大于效益,不具有使用价值时,或日本厚生劳动省规定的其他情形时,其会对该药品做出撤销上市批准的决定,新药的MAH须在收到再审查结果通知后立即将该药品撤出市场。日本新药再审查制度实施流程见
日本新药再审查制度已积累30余年的实施经验,形成了从制度目的、制度实施对象、制度实施方式到制度实施结果完整的制度体系。笔者通过提炼日本新药再审查制度每个实施环节的特点,对完善我国新药上市后再评价制度提出以下几点建议。
日本新药再审查申请资料主要包括使用情况调查资料、专项使用情况调查资料、上市后临床试验资料、ADR报告、文献研究报告、国内外对该药品所采取的措施报告以及严重药害事件报告,监测范围广。我国再注册申请资料中涉及新药监测的有药品临床使用情况及不良反应情况总结、Ⅳ期临床试验总结报告以及监测情况报告,建议对我国新药监测范围进行扩展,开展新药上市后用药的有效性调查以及特殊人群的用药调查。
日本对新药再审查申请资料采取文件调查、程序操作手册等实地调查以及科学性调查多种检查方式,保障再审查资料的真实性和完整性。完善我国再注册申请资料的检查方式,形成文件调查、实地调查和科学性调查3种检查方式结合的再注册申请资料检查体系,在保证文件所描述信息符合法定要求且真实的同时,发现潜在的科学性问题,以准确评估新药的安全性和有效性。
日本新药再审查制度具有鼓励新药创新,持续监测和评估药品风险效益平衡的双重功能,新药再审查期过后的3个月内,MAH必须提交再审查申请资料,接受厚生劳动省的再审查。根据《药品注册管理办法》《药品不良反应报告和监测管理办法》规定,我国新药监测期制度的目标是鼓励新药创新和持续监测药品风险效益;而5年一注册的再注册制度设计目标是定期评估已上市药品的风险效益。我国新药监测期制度及再注册制度的制度目标与日本新药再审查制度的制度目标一致,两者分别对应的是日本新药的再审查期和最终环节的再审查,但部分监测期为3或4年的新药在监测期过后需再过2年或1年才接受再注册。因此建议调整再注册期限,使其与监测期保持一致,监测期结束后立即开展对新药安全性、有效性重新评估的再注册,解决衔接问题。
The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}