中国科技论文统计源期刊 中文核心期刊
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖

 , 林铁豪, 谭昌成
, LIN Tiehao,, TAN Changcheng
, 林铁豪, 谭昌成
, LIN Tiehao,, TAN Changcheng
目的 建立离子色谱-电感耦合等离子质谱联用技术分析沉香化气丸中不同形态砷(As)和六价铬Cr(VI)。方法 以0.15 mol·L-1 硝酸溶液为萃取剂,80 ℃恒温箱中热浸提法提取沉香化气丸中砷甜菜碱(AsB)-二甲基砷酸(DMA)、三价砷As(Ⅲ)、砷胆碱 (AsC)、甲基砷酸(MMA)、五价砷As(V)等6种不同形态砷,磷酸氢二钾、磷酸二氢钾缓冲溶液与六水合氯化镁以及碱性提取液提取沉香化气丸中Cr(VI)。采用离子色谱-电感耦合等离子质谱直接测定相应含量。结果 沉香化气丸药材中砷的形态主要以As(Ⅲ)、As(V)为主,毒性极强的Cr(VI)含量极低,不同形态As回收率92.4%~105.8%,Cr(VI)回收率101.29%。结论 该方法灵敏,准确性好,是分离测试沉香化气丸中不同形态As和Cr(VI)的有效方法。
Objective To establish a method to analyze As-species and Cr(VI) in
按照《中华人民共和国药典》2015年版和国家药典委员会的要求,中药中有害元素控制项目主要包括铅、镉、汞、铜、砷等的总量。但有害元素的毒性并不直接取决于该元素的含量,而在于这些元素的化学形态以何种形式进入人体以及其生物有效性和被人体吸收与利用的程度。因此,用有害元素总量来评价中药毒性与风险存在很大缺陷。由于中药中有害元素属于痕量分析范畴,基质复杂且涉及的各形态含量更低,元素形态在分析过程中可能发生转变等,导致中药中有害元素形态分析成为中药质量控制中亟待解决的问题。
目前元素形态分析在环境科学和食品科学已取得了较大发展,常见技术为色谱与质谱技术联用。目前研究较多的有害元素主要包括砷(As)和铬(Cr)。如赵彤等[1]通过离子色谱和电感耦合等离子体质谱联机法测定稻米中砷的形态,贾娜等[2]采用离子色谱-电感耦合等离子体质谱法测定地下水中砷的4种形态。田雨荷等[3]采用反向离子色谱法测定水中痕量Cr(VI) 和Cr(Ⅲ),其他学者在采用色谱与质谱联用技术上也做了大量工作[4,5,6]。但是将色谱与质谱联用技术用于中药材、中药饮片以及中成药中有害元素测定的报道笔者较少见到 [7]。笔者在本实验中对沉香化气丸中不同形态As和Cr(VI)进行分析,以期为科学评价中成药中砷和铬限量提供参考。
iCAP Qc ICP/MS电感耦合等离子质谱仪 (美国赛默飞世尔科技有限公司),ICS5000离子色谱仪(美国赛默飞世尔科技有限公司),ME204电子天平(梅特勒-托利多公司,感量:0.1 mg)。
亚砷酸根标准溶液[As(III),含量:1 mg·L-1,批号:1707]、砷酸根标准溶液[As(V),含量:1 mg·L-1,批号:1707]、砷胆碱标准溶液[AsC,含量:1 mg·L-1,批号:1607]、砷甜菜碱标准溶液(AsB,含量:1 mg·L-1,批号:1707)、一甲基砷标准溶液(MMA,含量:1 mg·L-1,批号:1707)、二甲基砷标准溶液(DMA,含量:1 mg·L-1,批号:1707 )以及六价铬标准溶液[Cr(VI),含量:0.2 mg·L-1,批号:d1725072]均购自上海安谱实验科技股份有限公司。
砷标准溶液(批号:17041063,含量:1 mg·L-1),铬标准溶液(批号:17051163,含量:1 mg·L-1)购自国家钢铁材料测试中心钢铁研究总院;硝酸(优级纯,批号:UN2031,浓度:69%)购自默克公司;过氧化氢(分析纯,批号:20171030,浓度:30%)购自国药集团,水为本实验室自制去离子水,本实验中其他所用到的试药,除特别指出外,均为优级纯。
沉香化气丸样品来源于2016年国家基本药物评价性抽样,分别选取A公司和B公司部分样品。
分别移取6种砷标准储备液1.00 mL于10 mL塑料量瓶,用超纯水稀释至10.00 mL,摇匀待用。配成100 μg·L-1砷6种不同形态标准储备混合液。采用逐步稀释法,准确量取适量标准储备混合液于5 mL量瓶,配制成浓度为0,1,2,5,10 μg·L-1混合对照品溶液。
移取Cr(VI)标准储备液1.00 mL于10 mL塑料量瓶,超纯水稀释至10.00 mL,摇匀待用。配成100 μg·L-1Cr(VI)标准储备混合液。采用逐步稀释法,准确量取适量标准储备混合液于5 mL量瓶,配制0,0.5,1,2,5,10 μg·L-1混合对照溶液。
砷形态分析:取样品适量,粉碎,过内径180 μm筛(80目,《中华人民共和国药典》五号标准筛)。准确称取1.0 g经粉碎后的样品于50 mL塑料离心管,加入0.15 mol·L-1硝酸溶液20 mL,室温放置12 h,80 ℃恒温箱中热浸提1.5 h,每0.5 h振摇1 min。提取完毕,取出冷却至室温,采用8 000 r·min-1离心15 min,取上清液,经孔径0.45 μm有机滤膜滤过后进样测定。按同一操作方法作空白实验。
六价铬形态分析:准确称取样品0.5 g置于50 mL离心管,加入(磷酸氢二钾-磷酸二氢钾)缓冲溶液0.5 mL,六水合氯化镁0.4 g,加入碱性提取液(氢氧化钠、碳酸钠)2.5 mL,加水至25 mL,涡旋混合器上混匀。振荡器上振荡60 min,4 ℃下6500 r·min-1离心6 min。上清液用孔径0.45 μm滤膜滤过后进样测定。同一操作方法作空白实验。
总砷总铬分析:准确称取经粉碎后的样品0.5 g于聚四氟乙烯的消解罐中,加入硝酸5.0 mL,去离子水3.0 mL,设置程序升温程序,于微波消解仪中消解。冷却后转移并最后定容至50 mL量瓶,同法制备空白溶液。
As形态分析参数如下:色谱柱:AS7阴离子色谱柱(4 mm×250 mm);流动相:5 mmol·L-1碳酸铵(A)100 mmol·L-1碳酸铵(B),采用梯度洗脱方式,0~2.0 min,100%A;>2.0~5.5 min,100%B;>5.5~10 min,100%A,流速:1.0 mL·min-1,进样量:25 μL。
Cr(VI)分析参数,色谱柱:AG7阴离子色谱柱(4 mm×50 mm);流动相:0.76 mmol·L-1 硝酸铵(pH值9.3),流速:1.0 mL·min-1,进样量:25 μL。
2.4.1 标准曲线及其检出限 按照“2.3”项仪器条件,将“2.1”项所配制常见6种砷的化学形态对照溶液注入离子色谱仪进行分离后进入ICP-MS。依据响应值和浓度,得到相应的标准曲线图。检出限以3倍20次空白标准偏差响应值对应的浓度计算。由
表1 线性方程和检出限
Tab.1 Linear equation and detection limit
2.4.2 方法准确度 取沉香化气丸样品 6 份,加入砷混标溶液适量,按照“2.3”项仪器条件与供试品制备方法和条件进行萃取并测定。结果见
表2 沉香化气丸种6种不同形态砷的回收率实验结果
Tab.2
Results of recovery test on six different As-species in
由
2.5.1 标准曲线的绘制及检出限的确定 按照“2.3”项仪器条件,将“2.1”项所配制Cr(VI)对照溶液注入离子色谱仪进行分离后进入ICP-MS。依据相应值和浓度,得到相应标准曲线。检出限以3倍20次空白标准偏差响应值对应的浓度计算。
此时,得到Cr(VI)在0~10 μg·L-1浓度范围内满足线性方程
2.5.2 方法准确度 取沉香化气丸公司A样品 6 份,加入Cr(VI)对照溶液适量,由于样品中Cr(VI)含量极低,为减少实验误差,本实验Cr(VI)加入量按照10.0 μg·kg-1标准加入,并按照“2.3”项仪器条件与供试品制备方法和条件进行萃取并测定。结果表明该方法准确度较好。由
表3 沉香化气丸中Cr(VI)的回收率实验结果
Tab.3
Results of recovery test on Cr(VI) in
按照“2.2”项方法,提取沉香化气丸中AsB、DMA、As(Ⅲ)、AsC、MMA、As(V)等6种不同形态As。按照“2.3”项色谱条件,分别将“2.1”项混合对照品以及样品注入离子色谱-电感耦合等离子质谱仪,按照“2.5.1”项所得标准曲线法获得检测数据。典型色谱图见
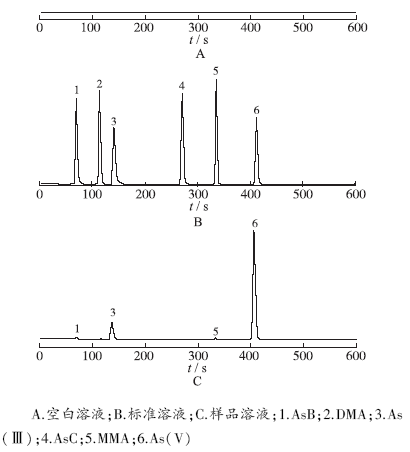
图1
3种溶液中6种不同形态砷色谱图
A.空白溶液;B.标准溶液;C.样品溶液;1.AsB;2.DMA;3.As(Ⅲ);4.AsC;5.MMA;6.As(V)
Fig.1
Chromatogram of six different As-species in three kinds of solution
A.blank solution;B.standard solution;C.sample solution;1.AsB;2.DMA;3.As(Ⅲ);4.AsC;5.MMA;6.As(V)
由
溶液通过离子色谱仪分离后,进入ICP-MS,记录信号强度,依据标准曲线,获得样品中砷含量,结果见
表4 样品中不同形态砷含量
Tab.4 Content of different As-species in samples μg·kg-1
由
按照“2.2”项方法,提取沉香化气丸中Cr(VI)。按照“2.3”项色谱条件,分别将“2.1”项Cr(VI)以及样品注入离子色谱-电感耦合等离子质谱仪,按照“2.5.1”项所得到的标准曲线法获得检测数据。典型色谱图见
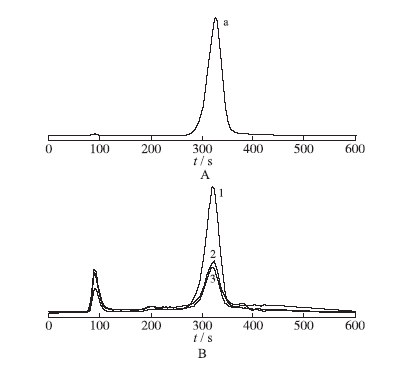
图2
标准溶液和供试品溶液中Cr(VI)色谱图
A.标准溶液;B.供试品溶液;a.Cr(VI) ;1.加标10 ppb样品;2.样品;3.空白
Fig.2
Chromatogram of Cr(VI) standard solution and sample solution
A.standard;B.rest solution;a.Cr(VI) ;1.10 ppb,add-standard;2.sample;3.blank
由
由
表5 样品中不同形态Cr含量
Tab.5 Content of different Cr-species in samples μg·kg-1
由
砷形态分析的难点是如何有效将目标物中的不同形态As提取出来。砷的化学形态主要有亚砷酸根[As(III)]、砷酸根[As(V)]、砷胆碱标准溶液[AsC]、砷甜菜碱[AsB]、一甲基砷标准溶液[MMA]、二甲基砷 [DMA]等。不同条件下容易相互转化。为确保不同形态As的萃取,对沉香化气丸中不同形态As的萃取条件进行优化具有极其重要的作用与意义。
3.1.1 萃取溶剂的选择 目前As的形态分析萃取溶剂主要有盐酸(0.12 mol·L-1)、硝酸(0.15 mol·L-1)等。盐酸较温和,多种形态砷化合物在该体系中发生相互转移和变化的可能性较小,具有较好的稳定性,但由于盐酸溶液中氯离子的大量存在,在质谱中会形成明显的多原子离子(ArCl -75),对砷(As-75 )离子有强烈的干扰。因此笔者在本实验采用硝酸提取方式。
采用硝酸提取时,如果硝酸浓度较高,容易氧化低价砷,导致砷化学形态变化,影响真实测定效果。因此需适度降低硝酸浓度,经考察,0.15 mol·L-1硝酸溶液适合沉香化气丸中不同化学形态砷的提取。
3.1.2 萃取温度的选择 分别比较70,75,80,85,90 ℃等5个萃取温度,发现提高温度对五价砷影响较大,当萃取温度低于85 ℃时,五价砷增长缓和。继续提高温度,五价砷增长较快。因此,萃取温度以<80 ℃为佳。笔者将该条件应用于菟丝子药材中不同形态砷的分析,亦获得类似的结果。
3.1.3 萃取时间的选择 分别选择30,60,80,90,100,120 min等6个时间段对样品萃取情况进行比较。萃取后,减压抽滤处理经过提取后的残渣。
按照传统微波消解方式对残渣进行消化处理并测定残渣中总砷含量来判断样品中As的萃取情况。结果表明,上述条件下萃取≥90 min,能有效将沉香化气丸中不同形态砷萃取完全。笔者将该条件应用于菟丝子药材中不同形态砷的分析,亦获得相同的结果。
中药中Cr(VI)萃取的难点就是尽可能保证Cr(VI)不转化为Cr(Ⅲ)。目前Cr(VI)形态分析的文献不多,大部分Cr(VI)的萃取主要采用1998年美国环境保护局(EPA)颁布的3060A法和浊点萃取法[8,9,10,11]。本实验采用美国环境保护局颁布的提取方法,用0.28 mol·L-1碳酸钠(Na2CO3)与0.5 mol·L-1氢氧化钠(NaOH)做萃取剂,90~95 ℃加热1 h。此时水溶性和非水溶性Cr(VI)全部提取完毕。由于该方法已经形成标准,故不再对其进行优化处理,笔者在本实验依据沉香化气丸特性,加入缓冲液(磷酸氢二钾—磷酸二氢钾缓冲溶液)和稳定剂(六水合氯化镁,)直接将美国环境保护局(EPA)方法应用于沉香化气丸中Cr(VI)的提取。
总之,笔者在本实验建立了IC-ICP-MS联用技术在沉香化气丸中测定AsB、DMA、As(Ⅲ)、AsC、MMA、As(V)等6种不同形态As和Cr(VI)。实验结果表明,测试分离度良好,方法准确可靠。通过本实验可知,沉香化气丸中砷、铬存在多种形态,而不同化学形态的生理毒性均不相同。因此,仅对样品中总砷和总铬含量进行测定不能有效反应样品毒性。建议国家食品药品监督管理局进一步强化有害元素的形态分析,制定更合理的重金属限度标准。
志谢:1.感谢赛默飞世尔科技(中国)有限公司王艳萍在本实验初期的指导;2.2016年我单位承担国家基本药物评价性抽验沉香化气丸和菟丝子的评价工作,两个品种的形态分析同步进行,在总结报告中共用部分色谱图
The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}