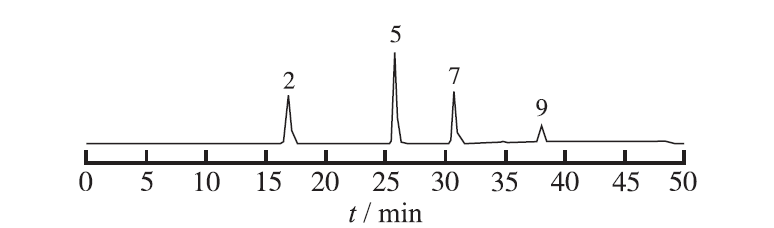
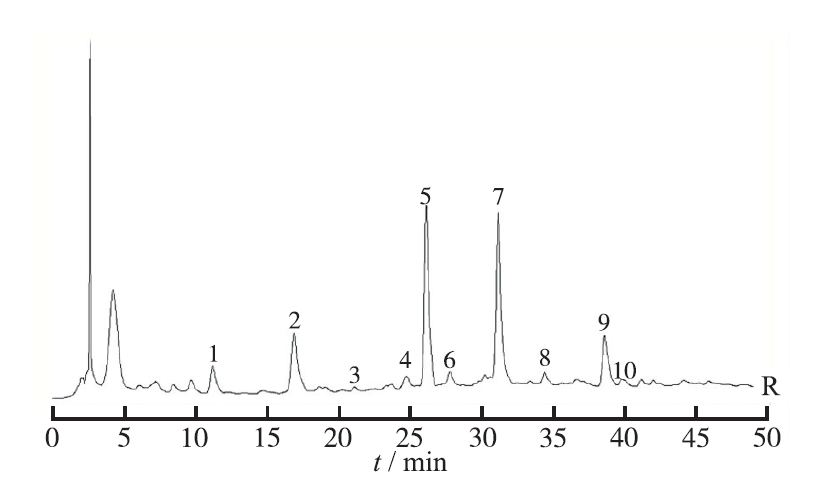
Objective To establish HPLC fingerprint and control the quality of chitosan Shanling Quban gels. Methods HPLC was applied to analyze the constituents of chitosan Shanling Quban gels. Using Agilent C18 Columns (4.6 mm×200 mm,5 μm). According to the National Pharmacopoeia Commission "HPLC Chromatographic Fingerprint Similarity Evaluation System" (2010 Edition), the chromatography was performed with methanol -0.1% phosphoric acid as mobile phase in gradient elution. The detection wavelength was 286 nm and the flow rate and column temperature was seted at 1.0 mL·min-1 and 30 ℃, respectively. Results The HPLC fingerprints of chitosan Shanling Quban gels were established, and 10 common characteristic peaks were labeled. The similarity of HPLC fingerprint was 0.9 for 10 batches of samples. Conclusion The HPLC fingerprint profile established in this experiment is highly specific and the method is simple and reliable which could reflect the quality of chitosan Shanling Quban gels.
with RSD of 1.46%. Conclusion: This method could be adopted as a standard for quality control of Quban gels and provide a reference for industrial production of this preparation.
目的 建立石荠苧黄酮提取物的指纹图谱,并测定5个指标成分的含量。方法 采用Hypersil BDS C18色谱柱(250 mm×4.6 mm,5 μm),以甲醇-0.1%磷酸水溶液为流动相,梯度洗脱,流速设为1.0 mL·min-1,柱温为30 ℃,检测波长270 nm。结果 得到9批次的石荠苧黄酮提取物HPLC指纹图谱,共标识了13个共有峰,其中5个为黄酮类化合物。各样品色谱图与对照色谱图的相似度均>0.96。木犀草苷、木犀草素、芹菜素、山奈酚和7-甲氧基汉黄芩素分别在19.42~194.2 μg·mL-1、0.520 0~5.200 μg·mL-1、0.214 0~2.140 μg·mL-1、0.134 4~1.344 μg·mL-1和0.013 60~0.136 0 μg·mL-1内线性关系良好,平均加样回收率分别为100.31%,98.94%,99.15%,100.95%和98.57%,RSD<3%(n=5)。结论 该方法稳定、可靠,可为石荠苧的质量评价提供参考依据。;OBJECTIVE To establish the method of fingerprint analysis and determination of flavonoids from Mosla scabra(MSF) by HPLC. METHODS HPLC separation was carried out on Hypersil BDS C18 (250 mm×4.6 mm, 5 μm) column with a gradient elution consisted of methanol and 0.1% phosphoric acid aqueous solution. The flow rate was kept at 1.0 mL·min-1. Column temperature was kept constantly at 30℃. The UV absorbance was monitored at 270 nm. RESULTS Nine batches of MSF were collected to generate representative chromatographic fingerprints. Thirteen common peaks were defined in characteristic fingerprints and five selected marker compounds in MSF were well identified by comparing the retention times of the reference standards. The similarity values of 9 samples were all higher than 0.96. Reasonable linear regressions of cynaroside, luteolin, apigenin, kaempferol and 7-methoxy wogonite were obtained in the fingerprints at ranges of 19.42-194.2 μg·mL-1, 0.520 0-5.200 μg·mL-1, 0.214 0-2.140 μg·mL-1, 0.134 4-1.344 μg·mL-1 and 0.013 60-0.136 0 μg·mL-1, respectively. Average recovery rates of five compounds were 100.31%, 98.94%, 99.15%, 100.95% and 98.57%, respectively, with the RSD of less than 3% (n=5). CONCLUSION The method is reproducible and precise and can be used for convenient quality assessment of Mosla scabra.

 , 熊鑫, 余南才
, 熊鑫, 余南才

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}