中国科技论文统计源期刊 中文核心期刊
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖

 , 王爽
, WANG Shuang
, 王爽
, WANG Shuang
目的 考察罗汉果苷V作为新型载体对难溶性药物的增溶作用。方法 以罗汉果苷V为载体,紫杉醇为模型药物,采用溶剂法制备紫杉醇-罗汉果苷V固体分散体。采用高效液相色谱(HPLC)法测定紫杉醇的含量,考察固体分散体中紫杉醇的饱和溶解度和体外溶出性能的变化。同时,用差示热扫描法(DSC)进行物相鉴别,评价药物在固体分散体中晶型变化。结果 紫杉醇-罗汉果苷Ⅴ固体分散体的饱和溶解度比紫杉醇增加约375倍;与紫杉醇比较,固体分散体的体外溶出速率和累积溶出度明显提高;差示热扫描法结果表明,紫杉醇在固体分散体内以无定形存在。结论 罗汉果苷V能明显增加难溶性药物的溶解度和体外溶出度,且该载体安全、无毒,有望成为难溶性药物增溶的新型载体。
Objective To investigate the solubilization effects of mogroside V as a novel carrier on the poorly soluble drugs. Methods Using mogroside V as a carrier and paclitaxel as a model drug, a solid dispersion of paclitaxel-mogroside V was prepared by the solvent method.The content of paclitaxel was determined by high performance liquid chromatography (HPLC). The saturated solubility and
口服给药简单,方便,患者依从性高,是药物递送的最优选途径。但目前超过40%市售药物、60%研发中药物都存在难溶性问题。得益于载体辅料的开发应用,改善疏水性药物的溶解性和生物利用度已有多种方法,如:使用磷脂复合物、生物相容性聚合物、固体分散体、脂质体或胶束等[1,2,3,4]。但目前满足上述方法要求、符合中药成分特性的辅料尤为缺乏。本课题组前期发现一些糖基化苷类化合物在水性环境中,能够形成与表面活性剂相似的结构,可作为固体分散体的载体,实现难溶性药物的高效增溶[5,6,7]。罗汉果甜苷是罗汉果的甜味成分。植物含量较高且水溶性好,目前已经有纯度98%以上成品用作食品添加剂,其甜度为蔗糖的300倍。罗汉果苷V是罗汉果甜苷的主要成分,是一种天然小分子化合物,由亲水性葡萄糖基和疏水性三萜类两部分组成,与已报道的糖苷类增溶载体结构相似,故选其为载体,以研究其对难溶性药物的增溶效果。罗汉果苷V甜度高,热稳定性好,色泽浅,易于使用,不受pH值(pH值在2~10)的影响。它在中国、美国、日本等一些国家作为非营养性天然甜味剂而闻名[8]。同时,含葡基辅料还可能通过与葡萄糖转运体1(glucose transporter 1,GLUT1)结合实现跨越血-脑屏障,进而实现对目标药物的脑靶向转运[9]。
紫杉醇(paclitaxel,PTX)是一种从短叶红豆杉(taxus brevifolia)茎皮中分离出来的复杂二萜类天然产物[10],由于能够与微管蛋白结合诱导细胞死亡并影响微管动力学[11],紫杉醇在治疗乳腺癌、卵巢癌和非小细胞肺癌方面表现出优异的抗肿瘤活性[12]。然而紫杉醇高度亲脂,水中溶解度低,研究报道显示其在小鼠中的生物利用度<3.6%[13],这一缺陷大大限制紫杉醇的临床应用。已上市产品紫杉醇注射液(Taxol©,商品名:泰素)使用乙醇和聚氧乙烯蓖麻油(50:50)的共溶剂系统来增加其溶解度,由于聚氧乙烯蓖麻油存在显著的毒副作用,如神经毒性、肾毒性、变态反应等[14],给患者的生命健康带来极大的危害。所以,新型载体或辅料的发掘,可为紫杉醇的临床制剂进一步开发提供帮助。
因此,笔者以罗汉果苷V为载体,以紫杉醇为模型药,采用溶剂法制备紫杉醇-罗汉果苷V固体分散体。考察其饱和溶解度和体外溶出特征,通过差示热扫描法(differential scanning calorimetry,DSC)考察紫杉醇在固体分散体中的物理存在形态。
LC-2010CHT液相色谱仪(含高压泵、柱温箱、自动进样器、紫外检测器、LC solution色谱工作站,日本岛津公司);KQ-250DE型数控超声波清洗器(昆山市超声仪器有限公司);BT125D电子天平(北京赛多利斯科学仪器有限公司,感量:0.01 mg);XW-80A旋涡混合器(海门市其林贝尔仪器制造有限公司);旋转蒸发装置(含N-1100型旋转蒸发仪、OSB-2100型油浴锅,上海爱朗仪器有限公司);SHB-Ⅲ循环水式多用真空泵(郑州长城科工贸有限公司);DKZ系列电热恒温振荡水槽(上海一恒科技有限公司);2XZ-2型旋片式真空泵(临海市永昊真空设备有限公司);差示扫描量热仪NETZSCH DSC 204(NETZSCH GERAETEBAU GmbH)等。
紫杉醇对照品(阿拉丁,含量:99%,批号:E1822047);罗汉果苷V(成都普瑞法科技开发有限公司,含量:98%,批号:PRF8093001);去离子水;乙腈为色谱纯;其他试剂为分析纯。
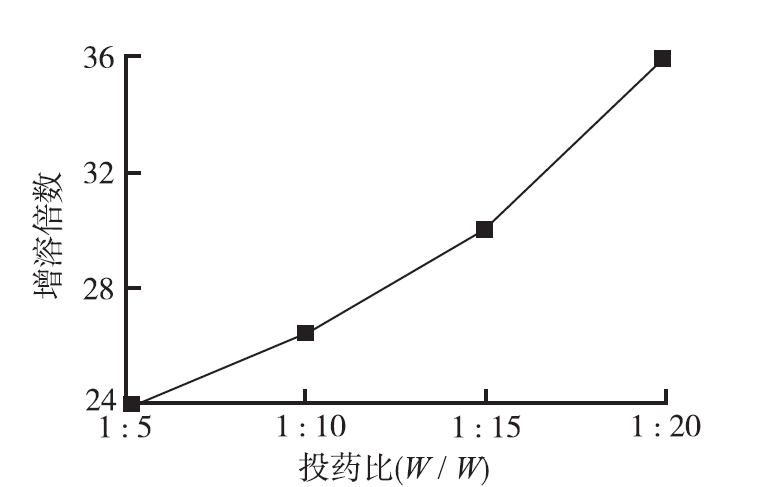
取紫杉醇适量,按1:5,1:10,1:15,1:20(
采用溶剂法制备紫杉醇-罗汉果苷V固体分散体,按1:20(
2.3.1 色谱条件 色谱柱:Inertsil©ODS-3(4.6 mm × 250 mm,5 μm);柱温:30 ℃;检测波长:227 nm; 流速:1 mL·min-1 ;流动相:乙腈-水(65:35)。
2.3.2 紫杉醇对照品溶液的配制 精密称定紫杉醇10 mg,置于10 mL棕色量瓶中,加入乙腈5 mL,超声使其溶解,用乙腈定容,配制得浓度约为1 mg·mL-1紫杉醇对照品溶液。
2.3.3 罗汉果苷V对照品溶液的配制 精密称定罗汉果苷V 10 mg,置于10 mL棕色量瓶中,加入乙腈5 mL,超声使其溶解,用乙腈定容,配制得浓度约为1 mg·mL-1罗汉果苷V对照品溶液。
2.3.4 紫杉醇-罗汉果苷V固体分散体溶液的配制 精密称定紫杉醇-罗汉果苷V固体分散体10 mg,置于10 mL棕色量瓶中,加入乙腈5 mL,超声使其溶解,用乙腈定容,配制得浓度约为1 mg·mL-1紫杉醇-罗汉果苷V固体分散体溶液。
2.3.5 专属性实验 分别取空白溶剂、罗汉果苷V对照品溶液、紫杉醇对照品溶液和紫杉醇-罗汉果苷V固体分散体溶液,按“2.3.1”条件,进行HPLC分析,紫杉醇典型色谱图见
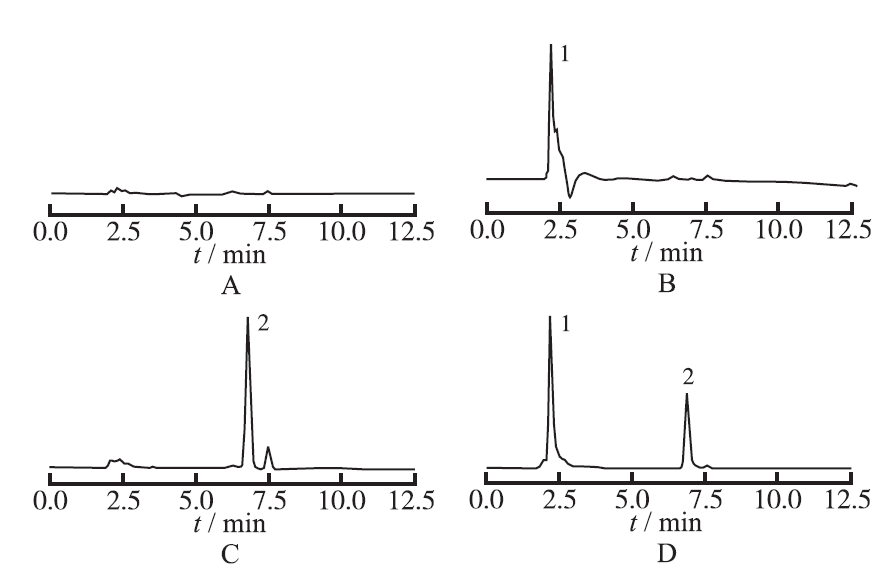
图1
4种溶液的HPLC图谱
A.空白溶剂;B.罗汉果苷V对照品溶液;C.紫杉醇对照品;D.紫杉醇-罗汉果苷V固体分散体溶液;1.罗汉果苷V;2.紫杉醇
Fig.1
HPLC chromatograms of four kinds of solution
1.blank solution;B.mogroside-V reference solution;C.paclitaxel reference solution;D.paclitaxel-mogroside-V SD solution;1.mogroside-V; 2.paclitaxel
2.3.6 检测限及定量限 取“2.3.2”项制备的紫杉醇对照品溶液,按一定比例稀释,得到一系列浓度溶液,进行HPLC分析,紫杉醇检测限(
2.3.7 线性关系考察 取“2.3.2”项制备的紫杉醇对照品溶液,按一定比例稀释,得到浓度分别为0.05,0.1,0.2,1,5,10,25,50 μg·mL-1的标准液,按“2.3.1”项条件进行HPLC分析,以紫杉醇峰面积(
2.3.8 精密度实验 精密量取“2.3.2”项制备紫杉醇对照品溶液0.5 mL,置于100 mL量瓶中,加流动相定容,得到5 μg·mL-1紫杉醇溶液。按“2.3.1”项方法重复测定6次,结果峰面积稳定,RSD为0.8%,表明仪器精密度良好。
2.3.9 重复性实验 取紫杉醇-罗汉果苷V固体分散体(1:20)约20 mg,共6份,精密称定。置于10 mL量瓶中,加适量乙腈溶解并定容。精密量取0.5 mL,置10 mL量瓶,加流动相定容。配制得相当于紫杉醇含量5 μg·mL-1溶液。按“2.3.1”项方法测定,计算6份样品中的紫杉醇含量。测定6份固体分散体中紫杉醇的含量分别为4.3%,4.2%,4.3%,4.4%,4.4%,4.3%,RSD为1.3%,表明该方法重复性良好。
2.3.10 稳定性实验 取“2.3.9”项下含紫杉醇5 μg·mL-1的紫杉醇-罗汉果苷V固体分散体溶液,室温下放置0,2,4,6 h,按“2.3.1”项方法分别测定,紫杉醇峰面积RSD为0.06%,表明紫杉醇溶液在室温下6 h内稳定。
2.3.11 加样回收率实验 参考文献[15]方法,分别精密称取紫杉醇适量,按处方比例(1:20)添加罗汉果苷V,分别制成高、中、低浓度溶液各3份,按“2.3.1”项方法测定。分析结果见
表1 紫杉醇加样回收率实验结果
Tab.1 Results of recovery test of paclitaxel
根据物理混合物饱和溶解度测定。取紫杉醇-罗汉果苷V不同比例的物理混合物(相当于紫杉醇1 mg),置于2 mL EP管中,加水0.5 mL,置于恒温振荡水浴锅中,37 ℃振摇饱和24 h,后取上清液,用孔径0.45 μm微孔滤膜滤过,取续滤液按“2.3.1”项方法测定紫杉醇浓度。结果如
分别取紫杉醇和紫杉醇-罗汉果苷V固体分散体(相当于紫杉醇1 mg),置于2 mL EP管中,加水0.5 mL,置于恒温振荡水浴锅中,37 ℃振摇饱和24 h,用孔径0.45 μm微孔滤膜滤过,取续滤液按“2.3.1”项方法测定紫杉醇浓度。结果显示紫杉醇的饱和溶解度约为0.1 μg·mL-1,紫杉醇/罗汉果苷V固体分散体的饱和溶解度约为37.5 μg·mL-1。与紫杉醇比较,固体分散体的饱和溶解度增加约375倍,表明罗汉果苷V明显增加紫杉醇的溶解度。
分别取紫杉醇、罗汉果苷V、紫杉醇与罗汉果苷V的物理混合物(1:20)、紫杉醇-罗汉果苷V固体分散体适量,置于铝制坩埚内,盖上盖子,密封。测试条件为:以空铝坩埚为参比,升温速率:10 ℃·min-1,扫描范围:40~300 ℃,结果见
参照文献[16]方法,以恒温振荡水浴锅为溶出装置,以pH值 6.8磷酸盐溶液(含0.2%聚山梨酯80)为溶出介质,每个溶出杯加入溶出介质100 mL,振荡频率50 r·min-1,温度为(37±0.5) ℃。称取等价于主药约2 mg的紫杉醇、物理混合物(1:20)、固体分散体分别加入溶出介质中,于5,10,20,30,60,120 min定时定位取样1 mL,经孔径0.45 μm微孔滤膜滤过,弃去初滤液。同时向溶出杯中补加新鲜介质1 mL。取出的样品立即按“2.3.1”项方法测定紫杉醇浓度,重复3次。结果见
累积溶出量(%)=(溶出的总物质量/投入量)×100%
溶出的总物质量=当前取样点介质浓度×介质体积+(之前取样点介质浓度×取样量)
笔者在本研究以食品添加剂罗汉果苷Ⅴ为固体分散体的载体,考察不同投药比例对紫杉醇增溶效果的影响。以药物增溶倍数为指标,筛选出最佳的固体分散体投药比例为1:20(
本研究的固体分散体处方简单、制备工艺简便,不仅显著提高紫杉醇的饱和溶解度,还有效改善其体外溶出性能。罗汉果苷V作为食品添加剂,安全、无毒。该研究结果表明罗汉果苷V有望成为新型载体辅料,在难溶性药物的制剂开发中具有极大应用价值。
The authors have declared that no competing interests exist.
| [1] |
The aim of this study was to improve the physicochemical properties and oral absorption of poorly water-soluble everolimus via preparation of a solid dispersion (SD) system using a solvent wetting (SW) technique. The physicochemical properties, drug release profile, and bioavailability of SD prepared by SW process were also compared to SD prepared by the conventional co-precipitation method. Solid state characterizations using scanning electron microscopy, particle size analysis and X-ray powder diffraction indicated that drug homogeneously dispersed and existed in an amorphous state within the intact polymeric carrier. Whereas, a film-like mass was obtained by a co-precipitation method and further pulverization step was needed for tabletization. The drug release from the SD tablet prepared by SW process at a ratio of drug to hydroxypropyl methylcellulose of 1:15 was markedly higher than the drug alone and equivalent to the marketed product (Afinitor , Novartis Pharmaceuticals), a SD tablet prepared by co-precipitation method, archiving over 75% the drug release after 30min. At the accelerated (40 C/75% R.H.) and stress (80 C) stability tests, the novel formula was more stable than drug powder and provided comparable drug stability with the commercially available product, which contains a potentially risky antioxidant, butylated hydroxyl toluene. The pharmacokinetic parameters after single oral administration in beagles showed no significant difference (P>0.01) between the novel SD-based tablet and the marketed product. The results of this study, therefore, suggest that the novel SD system prepared by the solvent wetting process may be a promising approach for improving the physicochemical stability and oral absorption of the sirolimus derivatives.
[本文引用:1]
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
DOI:10.1002/jps.22606
URL
[本文引用:1]
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
The purpose of this study was to prepare a functional dry powder capable of effectively improving the bioavailability of hydrophobic bioactive ingredients in crude drugs. A dry powder formulation from an ethanol extract of Brazilian green propolis was achieved in the presence of alpha-glycosyltransferase-treated stevia (Stevia-G). The resulting powder dispersed easily into an aqueous medium, and the average particle size of the suspension was about 350 nm. Propolis is known as a mixture of more than 200 ingredients; therefore, the suspension contains particles of hydrophobic compounds as well as dissolved molecules of several other compounds. In the suspension, the dissolved amounts of artepillin C and drupanin, representative hydrophobic bioactive compounds in propolis, in water were dramatically improved compared to their normal solubility. When the resulting propolis/Stevia-G powder was administrated into rats, pronounced absorption enhancement of artepillin C and drupanin could be detected. Stevia-G may have the potential to yield simultaneous absorption enhancement to multiple hydrophobic ingredients in crude drugs. (C) 2012 Elsevier B.V. All rights reserved.
[本文引用:1]
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}