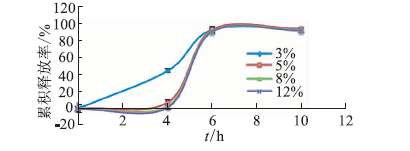
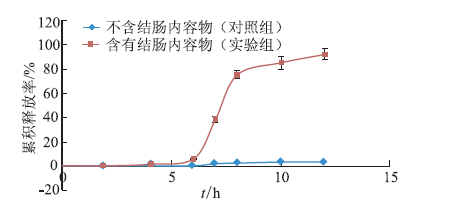
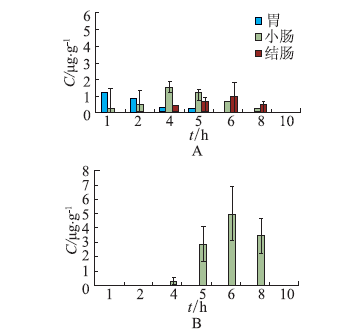
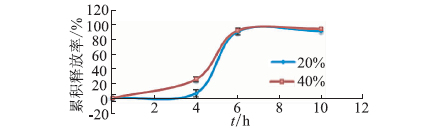
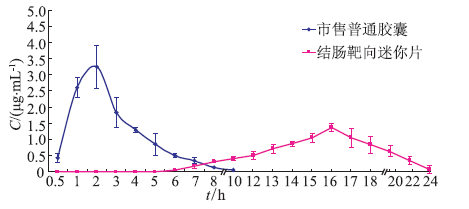
ObjectiveEnzyme triggered multi unit colon targeting mini tablet of indomethacin were prepared, in order to improve the target treatment of colon disease. MethodsDifferent proportion of enteric layer and chitosan layer were screened to optimize the prescription. The colon targeting mini tablets were prepared by direct compression method. The drug release properties were investigated in different release medium. Rats were used to investigate the distribution of tissue in vivo. The Beagle dogs were used to study the pharmacokinetics and bioavailability. ResultsThe optimum chitosan layer prescription: coating liquid concentration was 2%, plasticizer three citric acid ethyl ester (TEC) was 15%, an anti sticking agent amount of talc was 30%, coating weight was 5%; Enteric layer prescription: coating liquid solid content was 20%, plasticizer content of TEC was 5%, anti sticking agent talc powder dosage was 40%, coating weight was 3%. The chitosan multi unit colon targeted preparation seldom released in rat stomach and small intestine, released slowly in colon. The pharmacokinetics parameters in Beagle dogs were: Cmax=(3.25 + 0.672) mg·L-1, tmax= (2.00 + 0.014) h, AUC(0-∞) = (10.2 +0.871) mg·L-1·h, MRT (0-∞) = (2.82 + 0.180) h, CL= (2.46 + 0.202) L·h-1·kg-1. The release time of mini tablets for colon targeted was significantly prolonged and preserved stable blood concentration. ConclusionThe enzyme triggered multi unit colon targeting mini tablet of indomethacin showed good target to colon and sustained release effect, providing an important reference for the development of preparation of indomethacin for the treatment of colon disease.
口服结肠靶向给药系统(oral colon-specific drug delivery system,OCDDS),是一种新型的制剂技术,由于载体材料的特殊性质,可以直接将药物运送到结肠部位,避免在胃、小肠等胃肠道上端崩解或蚀解而释放药物。结肠靶向给药系统将药物运送到结肠处才开始崩解或蚀解并释放药物,可发挥局部或全身治疗作用[1-3]。根据载体材料释药的性质不同,OCDDS主要有pH值依赖型[4]、时间控制型[5]、压力控制型[6]和菌群/酶触发型[7]。其中菌群/酶触型OCDDS因其定位性好、可预测性高等特点,成为广大研究者关注的热点,是近年应用最为广泛的一种口服结肠靶向给药系统。吲哚美辛是最强的环氧化酶(COX)抑制药之一,主要用于急性风湿性及类风湿关节炎,并对结肠癌和直肠癌也有一定的治疗作用。但是吲哚美辛长期服用后药物不良反应(ADR)发生率较高,最常见的是胃肠道ADR,发生率为35%~50%[8-9]。通过先进的制剂手段改善吲哚美辛的溶解性,提高其体内生物利用度,并降低其对胃肠道的刺激,在结肠癌及其其他癌症的临床治疗上具有重要意义。
DASS, NGK.Colon-specific delivery of trsveratrol: optimization of multi-particulate calcium-pectinate carrier[J]. Int J Pharm, 2010, 385(1/2): 20-28.
[本文引用:1]
[2]
RAHMANS, ABDULLAHM, MASUM AA, et al.Formulation and evaluation of bi-layered sustained release matrix tablets of tramadol hydrochloride[J]. J Appl Pharm Sci, 2012,2(6): 129-134.
Abstract Bi-layer tablets of tramadol hydrochloride were prepared by direct compression technique incorporating an immediate release layer and a sustained release layer. An immediate release layer was successfully designed to release the bolus dose instantaneously. Water soluble Xanthan gum, water insoluble Kollidon SR and Eudragit L 100 were used as carriers in the sustained release layer of the matrix tablet. All the tablets were evaluated for thickness, diameter, weight variation, hardness and friability. The in vitro drug release was studied for eight hour, first two hours dissolution in acidic medium followed by six hour dissolution in buffer medium. Matrix tablet showed a sustained release rate with a controlled fashion as a function of the quantity of polymer used. The in vitro drug release data were fitted with several mathematical models and mean dissolution time along with fractional dissolution time values (T 25% , T 50% and T 80%) were calculated. Xanthan gum was found to be the most effective rate retarding agent compared to Kollidon SR and Eudragit L 100, when used at same ratio in the formulations.
ZHANGL, LIU XJ, ZHAI GX.Progress on oral colon-specific drug delivery system[J]. Chin J New Drugs Clin Remed, 2011, 30(8): 574-579.
Oral colon-specific drug delivery system(OCDDS)has gained increased importance for the delivery of drugs for the treatment of local diseases associated with the colon in recent years.In this article,the recent development of OCDDS was summarized,and in vitro and in vivo evaluation methods were also discussed.
RAO GS, MURTHYT.Formulation and evaluation of diltiazem HCl colon targeted tablets[J].Int J Res Pharm Chem, 2013,(3): 819-827.
[本文引用:1]
[5]
ZHANG JX, WU ZN, CHEN XJ.Optimization of preparation process for pH dependent-time Lag herba euphorbiae humifusae pellets for colon-specific delivery based on artificial neural network and particle swarm optimization algorithm[J]. Trad Chin Drug Res Clin Pharmacol,2012,23(1): 99-104.
Objective To optimize the pharmaceutical preparation process based on artificial neural network(ANN) and particle swarm optimization(PSO)algorithm.Methods Taking the pH dependent-time lag Herba Euphorbiae Humifusae pellets for colon-specific delivery as a research model,with weight increment of film coating and the proportion of plasticizer and film-forming material as the independent variables,and with overall desirability(OD) of in-vitro releasing property of the preparation as dependent variables,we optimized the process parameters with PSO algorithm by using back-propagation(BP)ANN modeling.Results The pellets prepared according to the optimized preparation process parameters had significant effect on colon-specific release in vitro.Conclusion The combination of BP ANN modeling with PSO provides an effective way of multi-dimensional optimization of complicated nonlinear systems involving pharmaceutical technology.
RUJIVIPATS, BODMEIERR.Improved drug delivery to the lower intestinal tract with tablets compression-coated with enteric/nonenteric polymer powder blends[J]. Eur J Pharm Biopharm,2010, 76(3): 486-492.
<p id="sp005">The objective of this study was to develop pH-erosion-controlled compression-coated tablets for potential colonic drug delivery with improved gastric resistance and pulsatile release based on compression-coatings of powder blends of the enteric polymer Eudragit® L100-55 and the extended release polymer ethylcellulose. Tablet cores containing model drugs of varying solubilities (acetaminophen, carbamazepine and chlorpheniramine maleate) were compression-coated with different ratios of Eudragit® L100-55:ethylcellulose 10cP FP at different compression forces and tablet core:compression-coat ratios. The compression-coated tablets were characterized by drug release, media uptake, erosion behaviour and wettability. All drugs were released in a pulsatile fashion in higher pH-media after a lag time, which was controlled by the erosion properties of the Eudragit L:ethylcellulose compression-coating. The addition of ethylcellulose avoided premature drug release in lower pH-media and significantly increased the lag time in higher pH-media because of a reduction in wettability, media uptake and erosion of the compression-coatings. Importantly, ethylcellulose also reduced the pH-dependency of the erosion process between pH 5.5 and 7.4. The lag time could also be increased by increasing the compression force and decreasing the core:compression-coat ratio. In conclusion, tablets compression-coated with blends of Eudragit L and ethylcellulose resulted in excellent release properties for potential targeting to the lower intestinal tract with no release in lower pH-media and rapid release after a controllable lag time in higher pH-media.</p>
GULBAKEA, JAIN SK.Chitosan: a potential polymer for colon-specific drug delivery system[J]. Exp Opin Drug Deliv, 2012, 9(6): 713-729.
There is an enormous growth and awareness of the potential applications of natural polymers for colon delivery of therapeutic bioactives. Chitosan (CH), a cationic polysaccharide, has a number of vital applications in the field of colon delivery and has attracted a great deal of attention from formulation scientists, academicians and environmentalists due to its unique properties.CH has been widely explored for the delivery of drugs, peptides, proteins and genes to the colon for different therapeutic applications. Sustained and controlled delivery can be achieved with CH-based formulations like CH-coated tablets, capsules, beads, gels, microparticles and nanoparticles. This review mainly focuses on various aspects of CH-based formulations, particularly development of colon-specific delivery of drug.The vital properties of CH make it a versatile excipient, not only for sustained/controlled release applications but also as biodegradable, biocompatible, bioadhesive polymer. The colon is recognized as the preferred absorption site for orally administered protein and peptide drugs. The main problem associated with CH is limited solubility at higher pH due to reduced cationic nature, which also reduces mucoadhesiveness. The application of newer targeting moiety with CH-based formulations for highly site-specific delivery of bioactive has to be evaluated for further improvement of therapeutic index (bioavailability).
PACIFICI GM.Differential renal adverse effects of ibuprofen and indomethacin in preterm infants: a review[J]. Clin Pharmacol,2014, 31(6): 111-116.
Abstract Objective The objective of this study was to evaluate the extent of renal adverse effects caused by ibuprofen or indomethacin in order to choose the safer drug to administer to preterm infants. Methods The following three parameters of renal function were taken into consideration: 1) the urine output; 2) the serum creatinine concentration; and 3) the frequency of oliguria. The bibliographic search was performed using PubMed and Embase databases as search engines. Results Urine output ranged from 3.5±1.2 to 4.0±1.4 mL/kg/h after ibuprofen treatment, and from 2.8±1.1 to 3.6±1.4 mL/kg/h after indomethacin treatment. The values for ibuprofen are significantly (P<0.05) higher than those for indomethacin. The serum creatinine concentrations ranged from 0.98±0.24 to 1.48±0.2 mg/dL after ibuprofen treatment, and from 1.06±0.24 and 2.03±2.10 mg/dL after indomethacin treatment. The values for ibuprofen are significantly (P<0.05) lower than those for indomethacin. The frequency of oliguria ranged from 1.0% to 9.6% (ibuprofen) and from 14.8% to 40.0% (indomethacin), and was significantly lower following ibuprofen than indomethacin administration. In infants with body weight lower than 1,000 g, oliguria appeared in 5% (ibuprofen) and 40% (indomethacin; P=0.02). Conclusion Indomethacin is associated with more severe renal adverse effects than ibuprofen. Ibuprofen is less nephrotoxic than indomethacin and should be used to treat patent ductus arteriosus in preterm infants. Immaturity increases the frequency of adverse effects of indomethacin.
PU YJ.Literature analysis of 137 ADR induced by indometacin oral preparation[J]. China Pharmacy, 2010, 21(36):3435-3436.
OBJECTIVE: To analyze the clinical features and correlation factors of ADR caused by Indometacin oral preparation and to provide references for rational use of drugs.METHODS: Retrieved from CBM Database,CNKI and Chinese Periodical Fulltext Database,literatures on ADR caused by Indometacin oral preparation during 1994锝2009 were analyzed.RESULTS: 137 ADR cases were enrolled.The number of male patients was more than female.Main manifestation of ADR were injury of digestive system(26 cases,18.98%),followed by general impairment of body(124 cases,17.52%),injurg of circulatory system(19 case,13.78%)and lesion of skin and its appendants(17 cases,12.40%).The situation of ADR was severe relatively.CONCLUSION: The occurrence of ADR causes by many factors.We should pay attention to f hemorrhage of digestive tract and shock.
KUSHWAHAP, FAREEDS, NANDA S et al. Design and fabrication of tramadol HCL loaded multiparticulate colon targeted drug delivery system[J]. J Chem Pharm Res, 2011(3): 584-595.
The aim of the present study is to develop a multiparticulate system containing pectin microspheres for the colon targeted delivery of Tramdol HCl (TMD) for the treatment of irritable bowel syndrome. This work combines pH-dependent solubility of shellac polymers and microbial degradability of pectin polymers. Pectin microspheres containing TMD were prepared by emulsion cross linking method using different ratios of TMD and pectin (1:2 to 1:5), stirring speeds (500-2000 rpm) and emulsifier concentrations (1.0% - 2.0% wt/vol). The yield of preparation and the encapsulation efficiencies were high for all pectin microspheres. Microspheres prepared by using drug: polymer ratio 1:3, stirring speed 1000 rpm, and 1.25% wt/vol concentration of emulsifying agent were selected as an optimized formulation. Shellaccoating of pectin microspheres was performed by oil-in-oil solvent evaporation method using coat: core ratio (5:1). Microspheres were evaluated for surface morphology, particle size and size distribution, swellability, percentage drug entrapment, and in vitro drug release in simulated gastrointestinal fluids (SGF). The release profile of TMD from Shellac-coated pectin microspheres was pH dependent. In acidic medium, the release rate was much slower; however, the drug was released quickly at pH 7.4. It is concluded from the present investigation that Shellac-coated pectin microspheres are promising controlled release carriers for colon-targeted delivery of TMD.
GADALLA HH,SOLIMAN GM,MOHAMMED FA,et al.Development and in vitro/in vivo evaluation of Zn-pectinate microparticles reinforced with chitosan for the colonic delivery of progesterone[J]. Drug Deliv,2016,23(7): 2541-2554.
Microporous bilayer osmotic tablet bearing dicyclomine hydrochloride and diclofenac potassium was developed using a new oral drug delivery system for colon targeting. The tablets were coated with microporous semipermeable membrane and enteric polymer using conventional pan-coating process. The developed microporous bilayer osmotic pump tablet (OPT) did not require laser drilling to form the drug delivery orifice. The colon-specific biodegradation of pectin could form in situ delivery pores for drug release. The effect of formulation variables like inclusion of osmogen, amount of HPMC and NaCMC in core, amount of pore former in semipermeable membrane was studied. Scanning electron microscopic photographs showed formation of in situ delivery pores after predetermined time of coming in contact with dissolution medium. The number of pores was dependent on the amount of the pore former in the semipermeable membrane. In vitro dissolution results indicated that system showed acid-resistant, timed release and was able to deliver drug at an approximate zero order up to 24 h. The developed tablets could be effectively used for colon-specific drug delivery to treat IBS. (C) 2011 Elsevier B.V. All rights reserved.
UR-REHMANT, TAVELINS, GROBNERG.Chitosan in situ gelation for improved drug loading and retention in poloxamer 407 gels[J]. Int J Pharm, 2011, 409(1/2) : 19-29.
A method for the in situ gelation of poloxamers and the mucoadhesive polymer chitosan has been developed by exploiting the tendency of poloxamer solution to form gel at physiological temperatures and of chitosan (CT) to form ionotropic gel structures in the presence of sodium tripolyphosphate (TPP). Novel poloxamer gels containing CT–TPP complex formed in situ during the administration were prepared by mixing poloxamer–CT and poloxamer–TPP solutions in double syringes. The micellization and gelation of poloxamer 407 in the presence of chitosan and/or TPP were studied using differential scanning calorimetry and tube inversion; both additives were found to reduce the critical micellization temperature and critical gelation temperature of poloxamer aqueous solution. The poloxamer gels containing CT–TPP complex formed in situ were found to exhibit reduced dissolution rate and superior release characteristics with three different drugs – metoprolol, doxycycline and flufenamic acid. Furthermore, by varying the compositions of the two solutions independently, it is possible to control the pH in a way to suit the solubilization of a drug as well as the specific environment of a particular application site. By varying the concentrations of chitosan, TPP and poloxamer, the delivery system can be fine-tuned to afford gels with specific properties, ranging from nanoparticle suspensions to semisolid gels. These in situ gels have the potential to increase the utility of thermo-reversible poloxamers in drug delivery.
Colon-specific delivery of trsveratrol: optimization of multi-particulate calcium-pectinate carrier
1
2010
... 口服结肠靶向给药系统(oral colon-specific drug delivery system,OCDDS),是一种新型的制剂技术,由于载体材料的特殊性质,可以直接将药物运送到结肠部位,避免在胃、小肠等胃肠道上端崩解或蚀解而释放药物.结肠靶向给药系统将药物运送到结肠处才开始崩解或蚀解并释放药物,可发挥局部或全身治疗作用[1-3].根据载体材料释药的性质不同,OCDDS主要有pH值依赖型[4]、时间控制型[5]、压力控制型[6]和菌群/酶触发型[7].其中菌群/酶触型OCDDS因其定位性好、可预测性高等特点,成为广大研究者关注的热点,是近年应用最为广泛的一种口服结肠靶向给药系统.吲哚美辛是最强的环氧化酶(COX)抑制药之一,主要用于急性风湿性及类风湿关节炎,并对结肠癌和直肠癌也有一定的治疗作用.但是吲哚美辛长期服用后药物不良反应(ADR)发生率较高,最常见的是胃肠道ADR,发生率为35%~50%[8-9].通过先进的制剂手段改善吲哚美辛的溶解性,提高其体内生物利用度,并降低其对胃肠道的刺激,在结肠癌及其其他癌症的临床治疗上具有重要意义. ...
Formulation and evaluation of bi-layered sustained release matrix tablets of tramadol hydrochloride
0
2012
Progress on oral colon-specific drug delivery system
1
2011
... 口服结肠靶向给药系统(oral colon-specific drug delivery system,OCDDS),是一种新型的制剂技术,由于载体材料的特殊性质,可以直接将药物运送到结肠部位,避免在胃、小肠等胃肠道上端崩解或蚀解而释放药物.结肠靶向给药系统将药物运送到结肠处才开始崩解或蚀解并释放药物,可发挥局部或全身治疗作用[1-3].根据载体材料释药的性质不同,OCDDS主要有pH值依赖型[4]、时间控制型[5]、压力控制型[6]和菌群/酶触发型[7].其中菌群/酶触型OCDDS因其定位性好、可预测性高等特点,成为广大研究者关注的热点,是近年应用最为广泛的一种口服结肠靶向给药系统.吲哚美辛是最强的环氧化酶(COX)抑制药之一,主要用于急性风湿性及类风湿关节炎,并对结肠癌和直肠癌也有一定的治疗作用.但是吲哚美辛长期服用后药物不良反应(ADR)发生率较高,最常见的是胃肠道ADR,发生率为35%~50%[8-9].通过先进的制剂手段改善吲哚美辛的溶解性,提高其体内生物利用度,并降低其对胃肠道的刺激,在结肠癌及其其他癌症的临床治疗上具有重要意义. ...
Formulation and evaluation of diltiazem HCl colon targeted tablets
1
2013
... 口服结肠靶向给药系统(oral colon-specific drug delivery system,OCDDS),是一种新型的制剂技术,由于载体材料的特殊性质,可以直接将药物运送到结肠部位,避免在胃、小肠等胃肠道上端崩解或蚀解而释放药物.结肠靶向给药系统将药物运送到结肠处才开始崩解或蚀解并释放药物,可发挥局部或全身治疗作用[1-3].根据载体材料释药的性质不同,OCDDS主要有pH值依赖型[4]、时间控制型[5]、压力控制型[6]和菌群/酶触发型[7].其中菌群/酶触型OCDDS因其定位性好、可预测性高等特点,成为广大研究者关注的热点,是近年应用最为广泛的一种口服结肠靶向给药系统.吲哚美辛是最强的环氧化酶(COX)抑制药之一,主要用于急性风湿性及类风湿关节炎,并对结肠癌和直肠癌也有一定的治疗作用.但是吲哚美辛长期服用后药物不良反应(ADR)发生率较高,最常见的是胃肠道ADR,发生率为35%~50%[8-9].通过先进的制剂手段改善吲哚美辛的溶解性,提高其体内生物利用度,并降低其对胃肠道的刺激,在结肠癌及其其他癌症的临床治疗上具有重要意义. ...
Optimization of preparation process for pH dependent-time Lag herba euphorbiae humifusae pellets for colon-specific delivery based on artificial neural network and particle swarm optimization algorithm
1
2012
... 口服结肠靶向给药系统(oral colon-specific drug delivery system,OCDDS),是一种新型的制剂技术,由于载体材料的特殊性质,可以直接将药物运送到结肠部位,避免在胃、小肠等胃肠道上端崩解或蚀解而释放药物.结肠靶向给药系统将药物运送到结肠处才开始崩解或蚀解并释放药物,可发挥局部或全身治疗作用[1-3].根据载体材料释药的性质不同,OCDDS主要有pH值依赖型[4]、时间控制型[5]、压力控制型[6]和菌群/酶触发型[7].其中菌群/酶触型OCDDS因其定位性好、可预测性高等特点,成为广大研究者关注的热点,是近年应用最为广泛的一种口服结肠靶向给药系统.吲哚美辛是最强的环氧化酶(COX)抑制药之一,主要用于急性风湿性及类风湿关节炎,并对结肠癌和直肠癌也有一定的治疗作用.但是吲哚美辛长期服用后药物不良反应(ADR)发生率较高,最常见的是胃肠道ADR,发生率为35%~50%[8-9].通过先进的制剂手段改善吲哚美辛的溶解性,提高其体内生物利用度,并降低其对胃肠道的刺激,在结肠癌及其其他癌症的临床治疗上具有重要意义. ...
Improved drug delivery to the lower intestinal tract with tablets compression-coated with enteric/nonenteric polymer powder blends
1
2010
... 口服结肠靶向给药系统(oral colon-specific drug delivery system,OCDDS),是一种新型的制剂技术,由于载体材料的特殊性质,可以直接将药物运送到结肠部位,避免在胃、小肠等胃肠道上端崩解或蚀解而释放药物.结肠靶向给药系统将药物运送到结肠处才开始崩解或蚀解并释放药物,可发挥局部或全身治疗作用[1-3].根据载体材料释药的性质不同,OCDDS主要有pH值依赖型[4]、时间控制型[5]、压力控制型[6]和菌群/酶触发型[7].其中菌群/酶触型OCDDS因其定位性好、可预测性高等特点,成为广大研究者关注的热点,是近年应用最为广泛的一种口服结肠靶向给药系统.吲哚美辛是最强的环氧化酶(COX)抑制药之一,主要用于急性风湿性及类风湿关节炎,并对结肠癌和直肠癌也有一定的治疗作用.但是吲哚美辛长期服用后药物不良反应(ADR)发生率较高,最常见的是胃肠道ADR,发生率为35%~50%[8-9].通过先进的制剂手段改善吲哚美辛的溶解性,提高其体内生物利用度,并降低其对胃肠道的刺激,在结肠癌及其其他癌症的临床治疗上具有重要意义. ...
Chitosan: a potential polymer for colon-specific drug delivery system
1
2012
... 口服结肠靶向给药系统(oral colon-specific drug delivery system,OCDDS),是一种新型的制剂技术,由于载体材料的特殊性质,可以直接将药物运送到结肠部位,避免在胃、小肠等胃肠道上端崩解或蚀解而释放药物.结肠靶向给药系统将药物运送到结肠处才开始崩解或蚀解并释放药物,可发挥局部或全身治疗作用[1-3].根据载体材料释药的性质不同,OCDDS主要有pH值依赖型[4]、时间控制型[5]、压力控制型[6]和菌群/酶触发型[7].其中菌群/酶触型OCDDS因其定位性好、可预测性高等特点,成为广大研究者关注的热点,是近年应用最为广泛的一种口服结肠靶向给药系统.吲哚美辛是最强的环氧化酶(COX)抑制药之一,主要用于急性风湿性及类风湿关节炎,并对结肠癌和直肠癌也有一定的治疗作用.但是吲哚美辛长期服用后药物不良反应(ADR)发生率较高,最常见的是胃肠道ADR,发生率为35%~50%[8-9].通过先进的制剂手段改善吲哚美辛的溶解性,提高其体内生物利用度,并降低其对胃肠道的刺激,在结肠癌及其其他癌症的临床治疗上具有重要意义. ...
Differential renal adverse effects of ibuprofen and indomethacin in preterm infants: a review
1
2014
... 口服结肠靶向给药系统(oral colon-specific drug delivery system,OCDDS),是一种新型的制剂技术,由于载体材料的特殊性质,可以直接将药物运送到结肠部位,避免在胃、小肠等胃肠道上端崩解或蚀解而释放药物.结肠靶向给药系统将药物运送到结肠处才开始崩解或蚀解并释放药物,可发挥局部或全身治疗作用[1-3].根据载体材料释药的性质不同,OCDDS主要有pH值依赖型[4]、时间控制型[5]、压力控制型[6]和菌群/酶触发型[7].其中菌群/酶触型OCDDS因其定位性好、可预测性高等特点,成为广大研究者关注的热点,是近年应用最为广泛的一种口服结肠靶向给药系统.吲哚美辛是最强的环氧化酶(COX)抑制药之一,主要用于急性风湿性及类风湿关节炎,并对结肠癌和直肠癌也有一定的治疗作用.但是吲哚美辛长期服用后药物不良反应(ADR)发生率较高,最常见的是胃肠道ADR,发生率为35%~50%[8-9].通过先进的制剂手段改善吲哚美辛的溶解性,提高其体内生物利用度,并降低其对胃肠道的刺激,在结肠癌及其其他癌症的临床治疗上具有重要意义. ...
Literature analysis of 137 ADR induced by indometacin oral preparation
1
2010
... 口服结肠靶向给药系统(oral colon-specific drug delivery system,OCDDS),是一种新型的制剂技术,由于载体材料的特殊性质,可以直接将药物运送到结肠部位,避免在胃、小肠等胃肠道上端崩解或蚀解而释放药物.结肠靶向给药系统将药物运送到结肠处才开始崩解或蚀解并释放药物,可发挥局部或全身治疗作用[1-3].根据载体材料释药的性质不同,OCDDS主要有pH值依赖型[4]、时间控制型[5]、压力控制型[6]和菌群/酶触发型[7].其中菌群/酶触型OCDDS因其定位性好、可预测性高等特点,成为广大研究者关注的热点,是近年应用最为广泛的一种口服结肠靶向给药系统.吲哚美辛是最强的环氧化酶(COX)抑制药之一,主要用于急性风湿性及类风湿关节炎,并对结肠癌和直肠癌也有一定的治疗作用.但是吲哚美辛长期服用后药物不良反应(ADR)发生率较高,最常见的是胃肠道ADR,发生率为35%~50%[8-9].通过先进的制剂手段改善吲哚美辛的溶解性,提高其体内生物利用度,并降低其对胃肠道的刺激,在结肠癌及其其他癌症的临床治疗上具有重要意义. ...

 , 敖惠, 王勇, 孙梦娟, 曹德英, 杜青
, 敖惠, 王勇, 孙梦娟, 曹德英, 杜青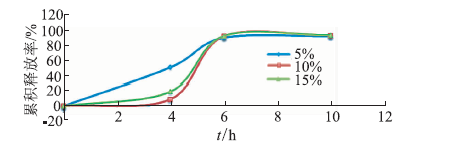


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}