中国科技论文统计源期刊 中文核心期刊
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖

 , 梅升辉
, MEI Shenghui
, 梅升辉
, MEI Shenghui
目的 评估高效液相色谱-质谱联用法(HPLC-MS/MS)测定人血浆伪人参皂苷GQ (PGQ) 浓度的不确定度。方法 全面分析HPLC-MS/MS法测定人血浆中PGQ浓度的整个过程,对测量重复性、标准品称量、工作液配制、生物样品配制、萃取回收过程、仪器允差和标准曲线拟和等引起的不确定度分别进行评定,计算合成不确定度并进行扩展。结果 人血浆低浓度(15.16 ng·mL-1 )、中浓度(2 516.67 ng·mL-1) 和高浓度(3 902.00 ng·mL-1)PGQ的扩展不确定度分别为1.39,177.74和262.69 ng·mL-1 (
Objective To evaluate the uncertainty of the pseudo-ginsenoside GQ (PGQ) concentration in human plasma by HPLC-MS/MS. Methods The whole process of PGQ determination by HPLC-MS/MS in human plasma was evaluated and the uncertainty caused by repeatability, weighing, standard solution preparation, biological sample preparation, extraction recovery process, recovery, instrument precision and calibration curve fitting were evaluated, respectively. The combined and expanded uncertainty values were both calculated. Results The expanded uncertainty values for low (15.16 ng·mL-1), medium (2 516.67 ng·mL-1) and high (3 902.00 ng·mL-1) levels of PGQ were 1.39, 177.74 and 262.69 ng·mL-1, respectively (
不确定度是指由于测量误差的存在,对被测量值的准确性造成影响的程度。建立不确定度的评定方法,通过考察方法中测量不确定度各分量的来源及大小,不仅可提高检测结果之间的可比性,更重要的是还可以改进实验方案,提高检测质量[1]。人参皂苷(ginsenoside)是人参的主要活性成分,对心血管系统、神经系统、免疫系统和生殖系统等方面均具有不同程度的保护作用[2]。伪人参皂苷GQ(pseudo-ginsenoside GQ,PGQ)注射液是国家一类新药(化学药品1.1类),其主要成分PGQ为奥克梯隆型人参皂苷,是以人参中20(
API 4000三重串联四极杆质谱仪,SHIMADZU LC-20AD液相色谱系统,Mettler AX105DR型电子天平,DB-3D型氮气吹干仪,Milli Q型纯水机。
PGQ (含量:98.2%,由吉林华康药业股份有限公司提供),内标为PGQ结构类似物人参皂苷Rc (含量:98.4%,由吉林华康药业股份有限公司提供),色谱纯甲醇购自Burdick & Jackson公司(HPLC级,批号:AH230-4),乙酸乙酯(分析纯,纯度:98.0%,批号:9282-03)和乙酸铵(分析纯,含量:98.0%,批号:20080126)购自北京化学试剂公司,去离子水由Milli Q纯化系统制备。
色谱柱使用Agilent公司的Poroshell 120 EC C8(2.1 mm×50 mm,2.7 μm)色谱柱,液相分离采用等度洗脱,每一个样品分析用时2 min,流动相:甲醇-10 mmol·L-1乙酸铵=(9∶1);流速0.3 mL·min-1;柱温40 ℃;进样量10 μL。
采用电喷雾离子源(electrospray ionization,ESI),在负离子电离模式下,选用多反应监测(multiple reaction monitoring,MRM)的质谱扫描方式进行测定,每个MRM通道的扫描时间为200 ms,PGQ和Rc(内标)的母、子离子对的质荷比分别为:799.7→161.1和1 077.8→191.0,其保留时间分别为0.57,0.53 min。
取PGQ对照品10.45 mg精密称定,以甲醇定容到10 mL量瓶,得到1.00 mg·mL-1标准品母液。准确吸取母液2.45 mL到5 mL量瓶,甲醇定容得500 μg·mL-1标准曲线工作液W。
取PGQ对照品10.40 mg精密称定,使用甲醇定容到10 mL量瓶,得1.00 mg·mL-1质控母液。分别准确吸取1.96,1.22 mL PGQ质控母液转移至5 mL量瓶,以甲醇定容至刻度,得到400 μg·mL-1质控工作液W1和250 μg·mL-1质控工作液W2。
精密称量内标10.23 mg于10 mL量瓶,甲醇定容得1.00 mg·mL-1内标储备液;移液管量取储备液4.95 mL到50 mL量瓶,超纯水定容得100 μg·mL-1内标工作液。
2.3.1 血浆标准曲线样品的配制 空白血浆来源于北京协和医院临床药理研究中心的志愿者。准确吸取工作液W 0.1 mL到10 mL量瓶,血浆定容得5 000 ng·mL-1S8;取S8溶液2 mL定容到5 mL量瓶得S7;取S8溶液1 mL定容到5 mL量瓶得S6;取S8溶液0.5 mL定容到5 mL量瓶得S5;取S6溶液0.5 mL定容到5 mL量瓶得S4;取S5溶液0.5 mL定容到5 mL量瓶得S3;取S4溶液0.5 mL定容到5 mL量瓶得S2;取S3溶液1 mL定容到10 mL量瓶得S1。血浆标准曲线S1~S8的浓度分别为5,10,50,100,500,1 000,2 000,5 000 ng·mL-1。将血浆的标准曲线样品进行分装,冻存于-80℃冰箱。
2.3.2 质控样品的配制 准确吸取质控工作液W1 0.1 mL到10 mL量瓶,血浆定容得4 000 ng·mL-1血浆质控Q3;准确吸取工作液W2 0.1 mL到10 mL量瓶,血浆定容得2 500 ng·mL-1血浆质控Q2;准确吸取Q2 1 mL到10 mL量瓶,血浆定容得250 ng·mL-1血浆溶液,然后从中吸取0.6 mL到10 mL量瓶,血浆定容得15 ng·mL-1血浆质控Q1。
2.4.1 血浆样品的制备 将血浆样品于室温下解冻,混匀10 s。加入25 μL内标工作液(100 μg·mL-1,去离子水溶解)于1.5 mL EP管中,加入血浆100 μL,涡旋振荡30 s,加入乙酸乙酯600 μL,混匀振荡2 min后,13 000 r·min-1离心10 min。取上层有机相500 μL转移至干净玻璃试管,于40 ℃ 氮气(N2)气流下吹干,并用复溶液(甲醇/水=50/50)100 μL复溶,振荡2 min,微型过滤器过滤,10 μL进样测定,记录质量色谱图及化合物的峰面积,采用内标法计算PGQ浓度。
2.4.2 质控样品的制备 质控样品浓度分别为15,2 500和4 000 ng·mL-1,制备方法同“2.3.1”项。
采用LC-MS/MS法进行检测,检测离子对为:PGQ为
标准曲线:
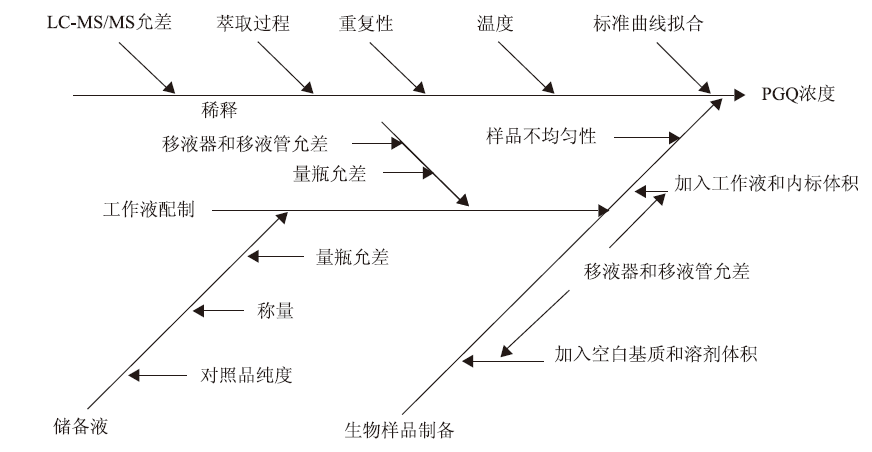
根据实验流程和检测方法,测量不确定度的来源主要包括重复性、称量、工作液配制、样品制备、萃取过程、仪器允差、曲线拟合、对照品纯度、温度和样品不均匀性等因素(
3.2.1 温度对测定的影响 实验室温度控制在(20±2)℃,PGQ和内标在相同温度下测定,温度引入的测量不确定度可忽略不计。
3.2.2 重复性引起的不确定度(用A类评定程序) 血浆质控低浓度15 ng·mL-1 (L),中浓度2 500 ng·mL-1 (M)和高浓度4 000 ng·mL-1 (H)样品共3组(
表1 样品重复测定数据
Tab.1
Results of the repeated determination of the sample
以每组测定结果的平均值表示测量结果,则低浓度平均值的标准偏差为:
计算得
3.2.2 天平称量PGQ及内标时引入的测量不确定度 (用B类评定程序) 减重法称取PGQ 10.45 mg,内标10.23 mg,称量引起的不确定度为:
天平的重复性误差
自动调零作为一次扣皮,则
不考虑重复性误差时,PGQ(
将称量引起的不确定度进行合成,则称量引起的相对标准测量不确定度为:
3.2.4 对照品溶液配制引入的测量不确定度 (用B类评定程序)
①量瓶引入的测量不确定度 实验所用A级量瓶(
②移液管引入的测量不确定度 实验所用A级移液管(
③加样枪引入的测量不确定度 实验所用Eppendorf加样枪型号有:20~200 μL(
④对照品溶液配制引入的测量不确定度 对照溶液稀释(包括储备液配制)共使用5 mL量瓶7次,10 mL量瓶3次,0.1 mL移液管1次,0.5 mL移液管4次,1 mL移液管2次,2 mL移液管1次,5 mL移液管1次;内标配制使用10 mL量瓶1次,50 mL量瓶1次,5 mL移液管1次,则溶液配制时PGQ和内标(IS)的相对标准测量不确定度为:
⑤含药标准样品和质控样品配制引入的测量不确定度(用B类评定程序) 配制标准含药样品(S)和质控样品(QC)所用加样枪的型号和次数相同:20~200 μL吸取25 μL 1次和100 μL 2次,100~1 000 μL吸取500 μL 2次。则配制标准含药样品时的相对标准测量不确定度为:
3.2.5 萃取过程 (用B类评定程序) 萃取过程引入的不确定度主要考虑回收率(RE)。配制3个浓度质控样品。同时用流动相配制与这3个质控浓度相同的样品,回收率=提取样品中的PGQ峰面积(A)/标准溶液中的PGQ峰面积(B),每组平行采样5次,则回收率的相对标准测量不确定度为(数据见
表2 PGQ的萃取回收率
Tab.2
Extraction recovery of PGQ
3.2.6 仪器量化引入的测量不确定度 所用质谱为API 4000三重串联四极杆质谱仪,所有液相为SHIMADZU LC-20AD液相色谱系统,质谱仪定量和液相仪吸样的最大允差为3%和1%,按均匀分布,则仪器量化的相对标准测量不确定度为:
3.2.7 线性拟合过程引入的测量不确定度(用B类评定程序) 测定8个不同浓度对照品血浆,用PGQ峰面积与内标峰面积的比值对PGQ浓度进行线性拟合(
表3 PGQ与内标峰面积比
Tab.3 Area ratio of PGQ with the internal standard
表4 各拟合标准曲线的参数
Tab.4 Parameters of the calibration curves
表5 拟合曲线计算出各对照品血浆的浓度结果
Tab.5 Back-calculated concentration of PGQ in standard plasma samples ng·mL-1
对照品血浆每个浓度重复分析5次,
自相关方差为:
残余标准差为:
测定低、中、高浓度质控样品各15次,
则曲线拟合的相对标准测量不确定度为:
0.073/2 516.67=0.000 029
0.090/3 902.00=0.000 023
3.2.8 样品不均匀性引入的测量不确定度 样品均为液态,使用前充分混合,则由样品不均匀性引入的不确定度可忽略不计。
3.2.9 对照品纯度引入的测量不确定度 对照品未提供不确定度,视为真实值,则引入的不确定度视为零。
依据不确定度传播规律对各相对标准测量不确定度进行合成:
则PGQ质控样品的合成相对标准测量不确定度分别为:
PGQ质控样品的合成标准测量不确定度分别为:
用简易评定法,对应的置信概率
当置信概率
笔者评价了采用HPLC-MS/MS分析方法测定血浆PGQ浓度的不确定度,按照实验流程寻找不确定度的来源并进行了量化。根据文献报道,其不确定度可估计为正态分布(
生物样品的药物浓度分析过程较复杂,环境、人员、仪器、量具等都会对检验结果带来一定不准确因素[10],通过对分析测定过程中误差的来源及大小进行分析,便于优化方法和改进检测方案,使检测结果更准确可靠,提高检测结果的质量。
The authors have declared that no competing interests exist.

{kind=link}
{kind=link}