中国科技论文统计源期刊 中文核心期刊
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖

目的 研究右佐匹克隆-β-环糊精包合物的最佳工艺条件。方法 采用饱和水溶液法,以包合物收率、包合率及溶出率为考察指标,通过正交设计筛选出制备包合物的优选条件。结果 优选包合条件为:右佐匹克隆与β-环糊精摩尔比1:1,包合温度60 ℃,包合时间6 h。通过红外光谱法、X线衍射法、差示扫描热分析鉴定,证实右佐匹克隆与β-环糊精已形成包合物。结论 该工艺简单可行,可为右佐匹克隆新剂型的开发提供依据。
β-环糊精(β-cyclodextrin,β-CD)是由7个葡萄糖单体以环状束缚在一起、具有一定容量的稳定空腔结构,是一种亲水性物质,对碱、热和光均稳定,生产成本低,常被用作增溶剂、稳定剂和促渗剂等药用辅料,由于其独特的分子内空腔结构(空腔内径70~80 nm),常被用于分子包合技术掩盖其他稍小分子物质的怪味、苦味。右佐匹克隆(相对分子质量388.81)分子大小适中,非极性强,适于β-CD空腔嵌入,因此将右佐匹克隆制备成右佐匹克隆-β-CD包合物不仅可以大大增加右佐匹克隆水溶性,而且有望掩盖药物的苦味。
78-1 磁力加热搅拌器(国华电器有限公司);HH-60恒温水浴锅(国华电器有限公司);UV 2401PC 紫外-可见扫描仪(日本岛津公司);IRPrestige-21 红外分光光度计(日本岛津公司);DMAX-1200X 衍射仪(日本理学公司);DSC204差示扫描量热仪(德国NETZSCH公司);超声波清洗器(天津市瑞普电子仪器公司)。
右佐匹克隆(成都康弘制药有限公司,原料药批号:130501,含量:99.1%);β-CD(Aladdin试剂上海有限公司,批号:D1315035,含量:98%)。
2.1.1 测定波长的确定 取右佐匹克隆原料25.2 mg,精密称定,置50 mL量瓶,加丙酮2 mL溶解,加0.1 mol·L-1盐酸定容,摇匀,即得504 μg· mL-1右佐匹克隆标准溶液。用紫外分光光度计在200~600 nm波长范围内扫描。结果表明,右佐匹克隆在305 nm波长处有最大吸收,且β-CD 在此处无吸收,故选305 nm为测定波长。
2.1.2 标准曲线的绘制 精密移取“2.1.1”项右佐匹克隆标准溶液 0.84,1.26,1.68,2.52,3.36 mL,分别置100 mL量瓶,加 0.1mol·L-1盐酸稀释至刻度,摇匀,于305 nm波长处测定其吸光度(
2.1.3 精密度实验 精密量取“2.1.1”制得的项右佐匹克隆标准溶液1.25 mL,置100 mL量瓶,加0.1 mol·L-1盐酸溶液稀释至刻度,摇匀,于 305 nm波长处测
2.1.4 重复性实验 平行取已制备包合物各5份,置10 mL量瓶,分别加纯化水及0.1 mol·L-1盐酸至刻度,40 ℃水浴振摇 30 min使溶解完全。溶液经孔径0.45 μm滤膜滤过于305 nm处测
2.1.5 回收率实验 精密量取“2.1.1”项右佐匹克隆标准溶液1.25,1.67,2.50 mL置100 mL量瓶,加0.1 mol·L-1盐酸稀释至刻度,摇匀,于 305 nm处测定各自
表1
右佐匹克隆回收率实验结果
2.2.1 饱和水溶液法制备包合物 取定量β-CD精密称定,置小锥形瓶中,在恒温水浴锅中正交设定包合温度下加纯化水适量,于磁力搅拌器上搅拌,制得饱和溶液,50 mL水浴保温。取相应量右佐匹克隆,精密称定,加入适量丙酮溶解,再将此溶液在搅拌速率100 r·min-1缓慢搅拌(本实验量下约用时3 min)加入β-CD溶液中,恒温搅拌正交设定时间,置冰箱4 ℃冷藏24 h,抽滤,快速用0.1 mol·L-1盐酸适量洗涤沉淀(弃去滤液),40 ℃恒温干燥4 h,即得右佐匹克隆-β-CD包合物,过五号筛,依紫外分光光度法含量测定方法测定并计算包合物收率(
2.2.2 包合物收率、右佐匹克隆包合率及右佐匹克隆溶出率的测定 将所得包合物精密称定质量,按下式计算包合物收率(
2.2.3 正交实验优选包合工艺 根据预实验结果,选取右佐匹克隆与β-CD投料摩尔比(A)、温度(B)及包合时间(C)为考察因素,以包合物收率、包合率及溶出率为考察指标,按 L9(34)正交实验表设计实验方案,进行工艺优选。各考察指标评分,分数=(当前值-所有该项目测量值中最小值)/(所有该项目测量值中最大值-所有该项目测量值中最小值)。正交实验设计各因素水平见
表2 正交实验因素水平
表3 正交实验方案与结果
极差分析结果表明,利用饱和水溶液法制备右佐匹克隆-β-CD包合物的优选工艺条件为右佐匹克隆与β-CD的投料量摩尔比1:1、包合温度 60 ℃ 、搅拌6 h,影响包合的因素主要为右佐匹克隆与β-CD投料摩尔比(A)。
方差分析结果见
表4 正交实验结果方差分析
取右佐匹克隆3批,按优选工艺条件进行验证性实验,制得包合物右佐匹克隆的包合率分别为58.99%,70.20%,64.54%;对应水溶出率(溶解度)为15.36%(20.1 mg),18.17%(24.4 mg),16.75%(21.9 mg),包合物收率62.39%,68.96%,65.68%,包合物的载药量为24.62%,23.23%,25.50%。结果表明优选工艺条件比较稳定,重复性良好。
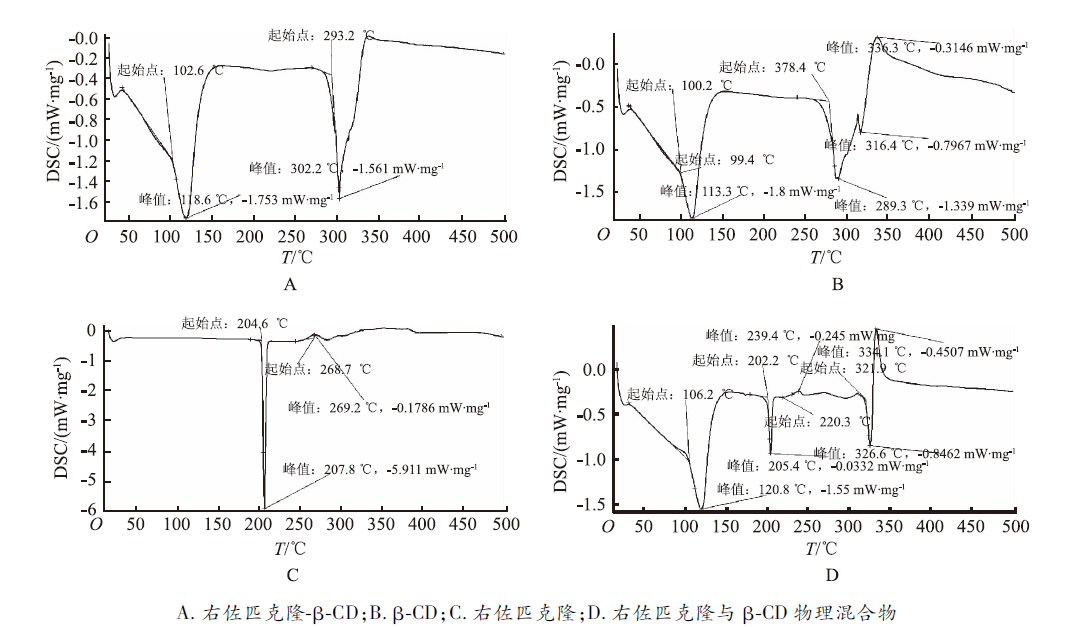
2.4.1 热分析法 采用差示扫描量热分析法(DSC),以氮气(N2)为保护气,升温速率10 ℃·min-1,氧化铝坩埚,温度范围 20~400 ℃。取包合物、β-CD、右佐匹克隆、物理混合物适量,用差示扫描量热分析仪测试热力学特征,结果见
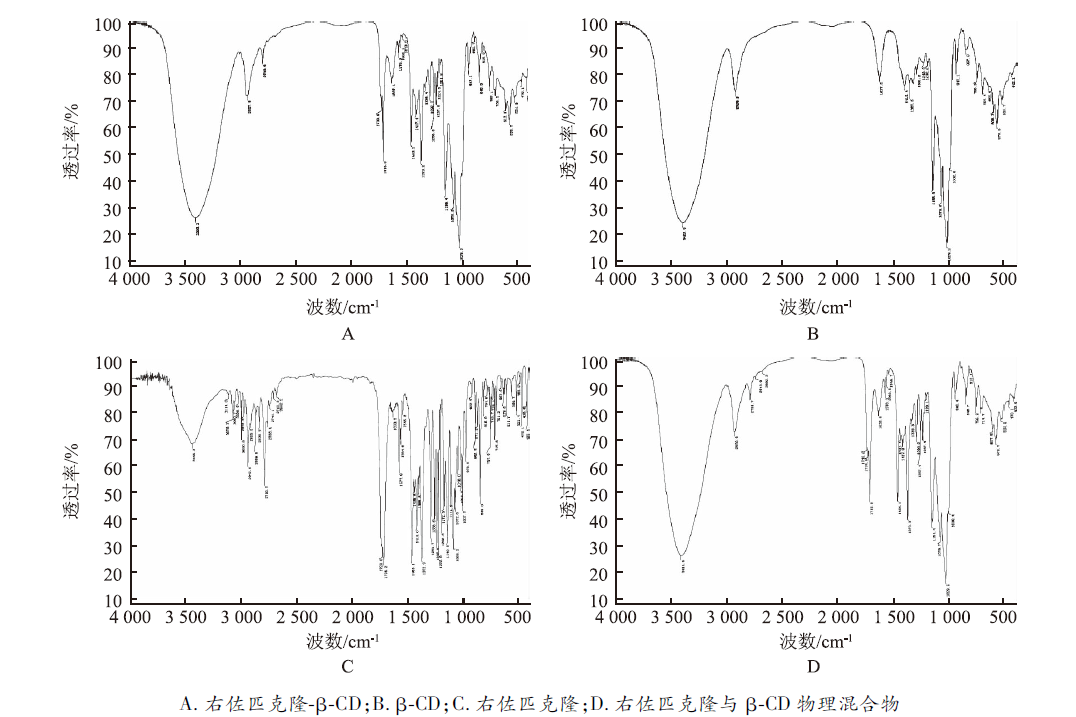
2.4.2 红外光谱法(IR) 从右佐匹克隆红外图谱可以看出,在 3 446.2 cm-1及 2 682.2~3 114.0 cm-1区域处表现出特征吸收峰。而包合物红外图谱在2 682.2~3 114.0 cm-1区域处许多特征吸收峰消失,仅2 927.8及2 790.9 cm-1处仍有特征吸收峰。而相同区域内物理混合物除在2 930.9及2 791.7 cm-1处有特征吸收峰外,在2 743.9及2 682.2 cm-1亦有特征吸收峰。且包合物与物理混合物 1 200~1 400 cm-1区域内的指纹吸收峰亦有差别,说明右佐匹克隆与β-CD主客分子间已形成新的物相(
2.4.3 X线衍射 对右佐匹克隆、β-CD、右佐匹克隆与β-CD物理混合物和包合物进行X射线衍射测定。测定条件:Cu靶;扫描范围为:2°~50°;扫描速度为:4°·min-1;电压:40 kV;电流:30 mA。扫描图结果见
本实验包合物经DSC法、IR法及X线衍射鉴别,确定已形成了右佐匹克隆-β-CD包合物。极差分析(
实验中考察包合率时用盐酸溶液为溶剂,因右佐匹克隆与佐匹克隆的溶解性相似[6],而佐匹克隆在水、乙醇中几乎不溶,在丙酮中略溶,在二氯甲烷、稀无机酸中易溶,故实验中选用盐酸溶液为溶剂考察包合率,以使药物从包合物中转出完全。用纯化水为溶剂考察包合物的溶解性,经微孔滤膜过滤除去可能残留未包合的药物。
本实验结果显示,利用饱和水溶液法制备右佐匹克隆-β-CD包合物的优选工艺条件为右佐匹克隆与β-CD的投料量摩尔比1∶1、包合温度 60 ℃ 、搅拌6 h。值得注意的是。升高温度似乎益于包合,与β-CD同佐匹克隆包合文献[8]不相一致,但与β-CD同其他药物包合的研究文献[9-10]近似,有待今后进一步考察。
由于右佐匹克隆在水中基本不溶,制备固体制剂难度大,需采用特殊工艺[11]或机械粉碎方法将右佐匹克隆粉碎到一定细度以保证该固体制剂口服后生物利用度(机械粉碎存在粉尘污染、材料损耗大,以及高活性右佐匹克隆粉雾吸入致操作人员快速催眠,易引发安全事故风险),右佐匹克隆与β-CD包合后,由于溶解性能改善,将大大降低固体制剂(如片剂)的生产难度。右佐匹克隆-β-CD的掩味效果还将为制作右佐匹克隆口腔崩解片、颗粒剂、口服液提供便利。
The authors have declared that no competing interests exist.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}