中国科技论文统计源期刊 中文核心期刊
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖

 , 赵庭波
, ZHAO Tingbo
, 赵庭波
, ZHAO Tingbo
目的 探讨杂环齐墩果酸衍生物-阿司匹林缀合物对血清素合成的抑制活性以及促骨形成活性。方法 采用高效液相色谱(HPLC)法、酶联免疫吸附测定(ELISA)试剂盒和荧光实时定量聚合酶链反应(PCR)测试杂环齐墩果酸衍生物-阿司匹林缀合物对RBL-2H3细胞中血清素合成的关键酶色氨酸羟化酶-1(TPH-1)的抑制率、TPH-1蛋白含量及mRNA的表达。构建去势大鼠骨质疏松症模型(OVX),并将骨质疏松大鼠随机分为给药组、甲状旁腺激素组、模型对照组和假手术组,给药35 d后采用HPLC法测定血清和小肠中血清素水平。采用噻唑蓝(MTT)法测定杂环齐墩果酸衍生物-阿司匹林缀合物对成骨细胞促进活性。结果 在RBL-2H3细胞中,杂环齐墩果酸衍生物-阿司匹林缀合物对TPH-1的抑制呈剂量依赖性,抑制率为20.4%~92.5%,缀合物7在10 μmol·L-1浓度有最高抑制率(92.5%),TPH-1蛋白含量为51 ng·mL-1,较对照组(216 ng·mL-1)显著降低,TPH-1mRNA表达也降低。与假手术组比较,模型对照组血清和小肠中血清素水平明显增高(
Objective To explore the effects of the heterocyclic oleanolic acid derivatives-aspirin conjugates on inhibition of serotonin biosynthesis and increase of bone formation. Methods The inhibition rate,content and mRNA expression of heterocyclic oleanolic acid derivatives-aspirin conjugates on tryptophan hydroxylase 1 (TPH-1),which was the principal enzyme in the biosynthesis of serotonin,were tested by HPLC,ELISA kit and real-time PCR,respectively.Osteoporosis model was established by ovariectomy (OVX).The rats were randomly divided into conjugate groups,parathyroid hormone group,model control group and sham operation group.Serum and gut serotonin levels were tested by HPLC after 35 days of administration,and the antiosteoporosis activity of the heterocyclic oleanolic acid derivatives-aspirin conjugates was evaluated by using osteoblast-like cells isolated from murine calvaria by MTT assay. Results The preliminary biological results showed that heterocyclic oleanolic acid derivatives-aspirin conjugates displayed a dose-dependent suppression on TPH-1 in RBL-2H3,and the inhibition rate was 20.4%-92.5%.Specifically,conjugate 7 at 10 μmol·L-1 concentration showed the greatest inhibition rate on TPH-1 (92.5%).Content of TPH-1 was 51 ng mL-1,significantly lower than that in the control group (216 ng·mL-1),as well as the TPH-1 mRNA.Compared with the sham operation group,levels of serum and gut serotonin were increased in OVX group,while they were significanlty decreased in conjugate groups,and reduced significantly in conjugate 7 group (232 and 155 ng·mL-1) as compared with OVX group (1 050 and 783 ng·mL-1); Moreover,heterocyclic oleanolic acid derivatives-aspirin conjugates could increase the bone formation. Conclusion This study provides information and basis for development of novel,efficient and harmfulless oleanolic acid derivatives with anti-osteoporosis.
齐墩果酸 (oleanolic acid, OA)又名庆四素,是一种齐墩果烷型五环三萜类化合物,以游离或与糖结合成苷的形式广泛存在于中草药,如夏枯草、女贞子、连翘、白花蛇舌草、丁香等[1-2]。齐墩果酸具有抗炎[3]、抗肿瘤[4]、抗菌[5]、抗人获得性免疫缺陷病毒[6]、降糖[7]等多种药理活性,且副作用小,毒性低,是一种具有开发潜力的先导化合物。通过初步研究,笔者发现齐墩果酸还能抑制肠源血清素5-羟色胺(5-HT)生物合成,而血清素进入血液循环后通过抑制骨细胞分化和增殖进而减少骨形成,并导致骨质疏松症[8-10]。因此,齐墩果酸是一类能够通过促骨细胞形成治疗骨质疏松症的新型药物,而目前临床上具有促进骨形成作用的药物只有甲状旁腺激素,但使用该药物却增加骨肉瘤发病率的风险[11]。然而,齐墩果酸对血清素合成的抑制作用仍然较弱,有必要对其进行结构修饰。因此,笔者将具有抗炎[12]、抗癌[13]、促骨形成[14-15]等多种药理活性的阿司匹林拼接到齐墩果酸上构建成一个单一的分子(缀合物1,
雌性SD大鼠,鼠龄10~11周,体质量205~255 g,雌性昆明乳小鼠,鼠龄1~3 d,体质量6~8 g;由武汉大学实验动物中心提供,实验动物使用许可证号:SYXK (鄂) 2008-0004;实验动物生产许可证号:SCXK (鄂) 2010-2012;动物级别:无特定病原体(SPF)级。在SPF级动物房自由饮水和摄食,每天光照12 h,室温20~25 ℃,相对湿度 (60±40)%。
齐墩果酸 (北京百灵威科技有限公司,批号:117093,含量>95%);阿司匹林 (北京百灵威科技有限公司,批号:DRE-C10024000,含量>95%)。齐墩果酸衍生物-阿司匹林缀合物由武汉大学刘远教授合成和结构确证 (含量>95%);达尔伯克改良伊格尔培养基(DMEM,Hyclone公司,批号:SH30022.01B)、α-MEM培养基 (Hyclone公司,批号:SH30024.01B)、胎牛血清 (Hyclone公司,批号:16250-078);色氨酸羟化酶-1(tryptophan hydroxylase 1,TPH-1)酶联免疫吸附测定(ELISA)检测试剂盒(武汉Cusabio公司);5-HT标准品(上海生物科技有限公司,批号:CSB-E13596h);甲状旁腺激素(parathyroid hormone,PTH,阿拉丁试剂有限公司,批号:P121395);二甲亚砜(DMSO)等其他试剂均为分析纯;RBL-2H3细胞株购自上海细胞典藏中心,本实验室冻存使用。
Agilent1200高效液相色谱仪 (安捷伦科技有限公司);BP211D型电子天平 (北京和悦达科技有限公司,感量:0.01 mg);BB16/BB5060仪器二氧化碳(CO2)培养箱(上海力创科学仪器有限公司);超净工作台(北京半导体设备一厂);ELx800通用酶标仪(美国BioTek公司);IX70-81FZ倒置显微镜(Olympus公司)。
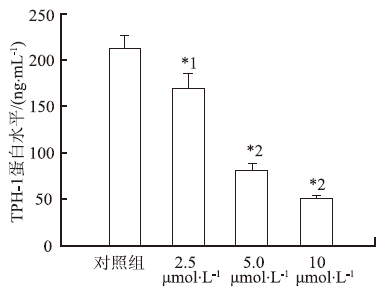
取对数生长期的RBL-2H3细胞株,悬浮于DMEM中,铺至48孔细胞培养板,在37 ℃、5%CO2培养箱中培养24 h后加入不同浓度的测试药物,终浓度分别为2.5,5和10 μmol·L-1,对照组仅加入溶剂DMSO (1 μL·mL-1培养液),培养48 h后弃去培养液,并用磷酸盐缓冲液(PBS)洗涤,然后每孔加入RIPA强裂解液100 μL裂解细胞,再加入PBS 200 μL稀释,离心后直接用HPLC法测定5-HT的峰面积,并计算出药物对5-HT生物合成的抑制率。抑制率 (%)=(1-测试化合物峰面积/对照组峰面积)×100%。
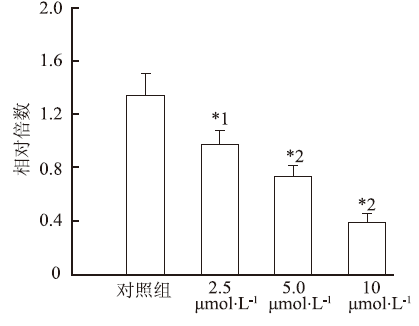
取对数生长期RBL-2H3细胞株悬浮于DMEM培养液中,铺至96孔板。待细胞完全贴壁后,弃去原培养液,加入含有测试药物的培养液100 μL,终浓度分别为2.5,5和10 μmol·L-1,在37 ℃、5%CO2培养箱中继续孵育48 h后,每孔加入5 mg·mL-1 MTT30 μL,继续孵育4 h,然后每孔加入DMSO 100 μL溶解。使用酶标仪在570 nm波长处测定每孔吸光度(
取对数生长期的RBL-2H3细胞株悬浮于DMEM培养液中,铺至24孔板,在37 ℃、5%CO2培养箱中孵育24 h后,加入测试药物,终浓度分别为2.5,5和10 μmol·L-1,对照组仅加入溶剂DMSO (1 μL·mL-1培养液),培养48 h后弃去培养液,并用PBS洗涤,然后将细胞反复的冻融,收集裂解液用TPH-1的ELISA试剂盒测试在450 nm波长处每孔
1.7.1 总RNA的提取 取对数生长期的RBL-2H3细胞株悬浮于含10%胎牛血清DMEM培养液中,铺至6孔细胞培养板中。待细胞完全贴壁后,加入不同浓度测试药物3 (对照组不加药物),培养2 d后弃去原培养液,每孔加入Trizol试剂1 mL,吹打细胞,使细胞充分裂解;随后加入三氯甲烷0.2 mL,室温静置5 min后离心15 min,将上层水相移置另一个1.5 mL无RNA酶的离心管中,并加入预冷的异丙醇0.5 mL沉淀RNA,离心后弃上清液,加入75%乙醇 (含0.1%DEPC,纯化水现配现用) 1 mL,用移液器吹打乙醇,洗涤RNA沉淀,离心后弃上清液,将离心管倒置,室温晾10 min后加入DEPC水80 μL溶解RNA。使用紫外分光光度计检测RNA浓度值,计算
1.7.2 实时荧光定量聚合酶链反应(PCR) 先登录GenBank database和文献查寻TPH-1基因序列[17],并确定所用引物序列为5'-GAAGACGTGGGGAGT TGTGT-3',5'-ACAGTGGAAAACACGGAAGG-3';接着以RNA为模板反转录出第一条cDNA链 (5×PrimeScriptTM Buffer (for Real Time) 2 μL; PrimeScriptTM RT Enzyme Mix I 0.5 μL; Oligo dT Primer (50 μmol·L-1) 0.5 μL; Random 6 mers (100 μmol·L-1) 2 μL; Total RNA 500 ng; 补加Rnase Free 蒸馏水至10 μL。设定程序为37 ℃ 15 min, 85 ℃ 5 s, 4 ℃ 5 min );随后以cDNA为模板进行Real-Time PCR (SYBR Premix Ex TaqTMⅡ 10 μL; PCR Forward Primer (10 μmol·L-1) 0.8 μL; PCR Reverse Primer (10 μmol·L-1) 0.8 μL; ROX Reference Dye (50×) 0.4 μL; DNA 模板2 μL; 重蒸水6 μL,设定程序为95 ℃ 30 s, 95 ℃ 5 s, 60 ℃ 30 s, 进行40个循环, 95 ℃20 s, 60 ℃ 60 s, 95 ℃ 15 s)。
构建去势大鼠骨质疏松症(osteoporosis in ovariectomized rats, OVX) 模型。将雌鼠分为给药组(剂量为20 mg·kg-1·d-1)、模型对照组、甲状旁腺激素组和假手术组,每组5只。在摘除卵巢造模术后第4天开始给药,甲状旁腺激素组腹腔注射甲状旁腺激素20 mg·kg-1·d-1,连续给药35 d后经颈动脉取血。将血液在37 ℃下静置45 min后得到血清,用HPLC法测定血清素含量,并用标准曲线定量。
无菌条件下将25只出生2~3 d乳小鼠处死,取出其头盖并刮去软组织,PBS清洗,剪碎后转移到培养瓶中,用混合酶 (0.25%胰酶+0.1%Ⅱ型胶原酶=1∶1),37 ℃消化15 min,弃去上清液;再用0.1%Ⅱ型胶原酶消化4次,合并上清液并用培养液终止消化,将上清液用孔径0.075 mm(200目)滤网滤过,2 000 r·min-1离心5 min收集细胞,将收集的细胞接种到培养瓶中培养4 d,洗去未贴壁的细胞,剩下细胞即为成骨细胞;取对数生长期成骨细胞悬浮α-MEM培养液中,铺至96孔板。待细胞完全贴壁后,弃去原培养液,加入浓度为10 μmol·L-1测试药物,对照组仅加入溶剂DMSO (1 μL·mL-1培养液),置于37 ℃、5% CO2培养箱中,培养12 h后弃去培养液,每孔加入5 mg·mL-1 MTT 30 μL,继续孵育4 h,然后每孔加入DMSO 100 μL溶解。使用酶标仪在570 nm波长测定每孔
采用SPSS17.0版统计软件进行分析,进行各组数据的正态性检验,对符合正态性分布的数据采用均数±标准差(
5-HT主要由TPH-1合成,因此,5-HT的水平变化反映TPH-1活性。通过应用高表达TPH-1的RBL-2H3细胞株来检测缀合物对5-HT生物合成的抑制率,并以对TPH-1有较好抑制剂活性的药物LP533401为阳性对照,结果见
表1 各种药物对5-HT生物合成的抑制率
Tab.1
Inhibitory effects of the test drugs on 5-HT biosynthesis %,
表2 各种药物对RBL-2H3细胞的毒性
Tab.2
Toxicity of the tested drugs on RBL-2H3 cells %,
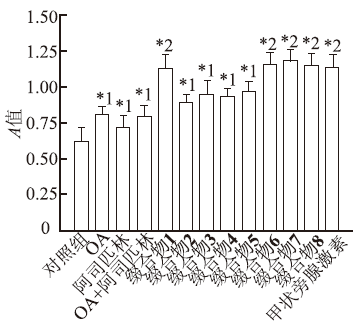
检测各种药物在浓度10 μmol·L-1时对TPH-1蛋白量的影响,结果见
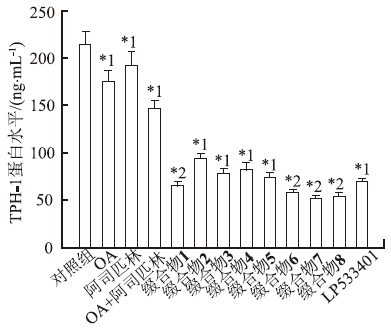
图2
不同药物作用后TPH-1蛋白量测定值(
与对照组比较,*1
Fig.2
Protein expression of TPH-1 after various treatment(
Compared with control group, *1
结果见
结果见
通过MTT法探讨齐墩果酸衍生物-阿司匹林缀合物的促成骨细胞增殖的活性,结果见
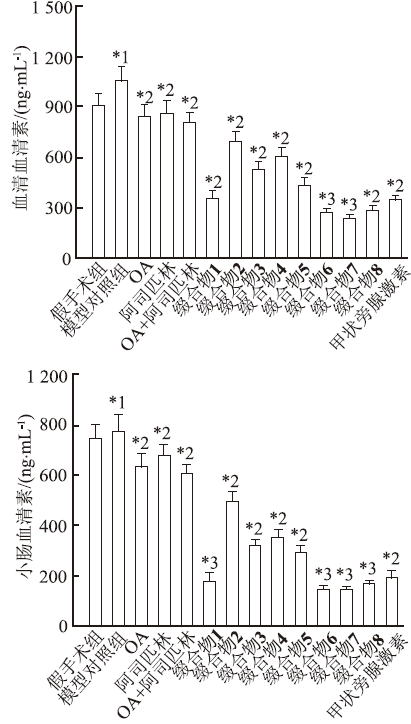
图5
不同药物作用后大鼠血清和小肠血清素水平(
与假手术组比较,*1
Fig.5
Serotonin level in serum and intestine of rats after various treatment(
Compared with sham operation group, *1
本研究结果显示,这些齐墩果酸衍生物-阿司匹林缀合物对血清素均有抑制作用,强于先导化合物OA和阿司匹林;与物理混合组相比,化学合成的缀合物对5-HT合成有更强抑制作用。但缀合物之间对5-HT合成的抑制作用差异有统计学意义,其中异卟恶唑、吡唑和吡嗪类齐墩果酸衍生物-阿司匹林缀合物 (缀合物3,4,5)抑制5-HT合成的活性低于缀合物1和阳性药物LP533401,而喹啉、吲哚和喹喔啉类齐墩果酸衍生物-阿司匹林缀合物 (缀合物6,7,8) 抑制血清素合成活性要强于缀合物1和阳性药物LP533401,尤其是缀合物7在10 μmol·L-1对血清素的抑制作用最强,分别是齐墩果酸和阿司匹林的13和20倍,这些结果说明苯并杂环类齐墩果酸衍生物-阿司匹林缀合物能提高母体化合物抑制血清素合成的活性。进一步研究发现,这些齐墩果酸衍生物-阿司匹林缀合物对RBL-2H3细胞株基本没有毒性。这说明缀合物对血清素合成的抑制活性不是其细胞毒性,而是其对TPH-1的抑制活性,并能明显抑制TPH-1蛋白含量及其mRNA表达,其中缀合物7也展现出最强的活性,强于阳性药物LP533401,并呈浓度依赖性。在动物水平上,与空白对照组比较,骨质疏松模型组大鼠血清和小肠中血清素水平明显增高,大鼠给予缀合物后血清素水平却明显降低,其抑制血清素的作用也强于齐墩果酸、阿司匹林和齐墩果酸与阿司匹林混合物。这些结果说明缀合物在动物体内也能够抑制血清素的合成。缀合物能显著促进成骨细胞增殖,尤其是缀合物7促成骨细胞增殖活性最强。
综上所述, TPH-1是治疗骨质疏松症的一个重要靶点,而齐墩果酸衍生物-阿司匹林缀合物能够抑制TPH-1,从而抑制血清素合成,促进成骨细胞的增殖。这为今后开发新型、高效、低毒的齐墩果酸类抗骨质疏松药物提供新的思路。
The authors have declared that no competing interests exist.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}