中国科技论文统计源期刊 中文核心期刊
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖

 , 马慕白
, MA Mubai
, 马慕白
, MA Mubai
丙戊酸是临床一线的广谱抗癫痫药物,但其代谢易受遗传、联合用药等多种因素影响,因此如何更好实施丙戊酸个体化给药,是临床备受关注的问题。群体药动学将经典的药动学原理与群体统计模型结合,定量地考察患者群体中药物浓度的影响因素,可更好表征个体间差异,实现个体化给药。近年来,许多研究报道丙戊酸的群体药动学研究及其在个体化给药中的应用。该文对近年来丙戊酸相关研究进行综述,总结联合用药及基因多态性对丙戊酸代谢的影响,重点阐述群体药动学指导下丙戊酸个体化给药的最新研究进展。
Valproic acid is a first-line broad-spectrum antiepileptic drug, however, the pharmacokinetics of valproic acid are affected by many factors, such as heredity, drug combination and so on. So, how to realize the individualized dosing regimen of valproic acid has received much attention. Population pharmacokinetics combines classical pharmacokinetic principles and population statistical models to quantitatively evaluate the influence factor of drug concentration in the patient population. Thus, it can optimize characterization of the differences among individuals. This article reviews the researches about valproic acid in recent years, and summarizes the effect of the factors on the metabolism of valproic acid, such as drug combination and gene polymorphism. Additionally, focus on the latest research progress of individual administration of valproic acid under the guidance of population pharmacokinetics.
[编者按] 癫痫是一种常见的神经系统疾病,我国癫痫患者约有900万例。丙戊酸是一线的广谱抗癫痫药物,对各型癫痫均有一定的治疗作用。但其治疗窗窄,体内代谢与药物疗效个体间差异大,因此如何实现其个体化给药是临床备受关注的问题。作者长期从事抗癫痫药物个体化给药研究,《丙戊酸个体化给药研究进展》一文,对近年来丙戊酸相关研究进行了综述,详细总结合并用药及基因多态性对丙戊酸代谢的影响,重点阐述群体药动学指导下丙戊酸个体化给药的最新研究进展,可为临床医生及从事癫痫个体化给药研究的学者提供有价值的参考和借鉴。
丙戊酸(valproic acid,VPA)是临床常用广谱抗癫痫药物,对全身性发作、部分性发作及特殊类型综合征等各型癫痫均有一定的治疗作用。此外,还可用于预防和治疗双相情感障碍的急性躁狂症状等[1]。目前,丙戊酸抗癫痫药理机制尚未完全阐明,但主要与γ-氨基丁酸(γ-aminobutyric acid,GABA)有关,该药能抑制脑内GABA转氨酶,减慢脑内抑制性神经递质GABA代谢,提高GABA浓度[2-3],并作用于突触后感受器部位,提高突触后膜对于GABA的反应性;此外还有研究指出,丙戊酸可通过抑制钠离子(Na+)通道和L型钙离子(Ca2+)通道发挥抗癫痫作用[4]。丙戊酸口服吸收迅速,生物利用度高,但其治疗窗窄,药动学参数具有明显的个体差异[5],为确保临床疗效,降低不良反应,应实施个体化给药方案。近年来丙戊酸的群体药动学(population pharmacokinetics,PPK)研究逐渐得到重视,国内外对影响丙戊酸给药剂量相关的基因多态性及PPK模型的研究不断深入。笔者对影响丙戊酸代谢的多种因素进行总结的同时,重点阐述PPK模型指导下丙戊酸个体化给药的国内外最新研究进展,为其临床个体化治疗提供理论依据。
肝脏是丙戊酸代谢的最主要脏器,约97%丙戊酸经过肝脏代谢,在体内的代谢途径主要有3种,见
目前有700多种药物对丙戊酸代谢有影响,20多种药物对丙戊酸的清除有重要影响,如阿司匹林、亚胺培南、拉莫三嗪、卡马西平等。
2.1.1 阿司匹林等非甾体类抗炎药(non-steroidal anti-inflammatory drugs,NSAIDs) 水杨酸类药物中阿司匹林血浆蛋白结合率高,可将丙戊酸从血浆结合蛋白上置换下来,从而增加体内游离型丙戊酸浓度,使治疗和毒性作用增加[12]。已有报道显示,当服用丙戊酸患儿合并使用阿司匹林时,游离型丙戊酸可增加4倍[13]。此种相互作用更易发生在大剂量和长期合用水杨酸类药物时,小剂量时不会造成重大影响。此外,丙戊酸可抑制体内血小板的聚集,与阿司匹林和双嘧达莫合用时,可降低血小板聚集,延长出血时间[12]。因此,当患者大剂量和长期联用水杨酸类药物时,应密切关注丙戊酸的毒性,注意检测血药浓度及血常规。
2.1.2 碳青霉烯类抗生素(carbapenem) 碳青霉烯类抗生素可显著降低丙戊酸血药浓度,并增加癫痫发作风险。目前,确切的相互作用机制尚未完全阐明。SUZUKI等[14]研究发现,乙酰肽水解酶(acetylpeptide hydrolases,APEH)可特异性地将VPA-G水解成丙戊酸。而碳青霉烯类抗生素可抑制APEH活性,从而显著降低丙戊酸的浓度[15]。临床已多次报道,丙戊酸合并使用亚胺培南、美罗培南和帕尼培南,可造成丙戊酸血药浓度低于有效治疗浓度下限[16-17]。尽管增加丙戊酸剂量亦不能使其血药浓度升高,但当停用碳青霉烯类抗生素时,丙戊酸浓度通常会快速升高[18]。提示,丙戊酸应尽量避免与碳青霉烯类抗生素联合使用,当癫痫患者已使用丙戊酸治疗时,应选择其他替代抗生素。必须联用时,应补充其他抗癫痫治疗,并密切关注丙戊酸血药浓度及药物反应[18-19]。
2.1.3 拉莫三嗪(lamotrigine) 丙戊酸联用拉莫三嗪时,可显著增加后者的血药浓度和发生潜在的严重威胁生命的皮疹风险,如Stevens-Johnson综合征、中毒性表皮坏死松解症[20-21],该机制主要是因为丙戊酸可竞争性抑制拉莫三嗪葡萄糖醛酸化。MAY等[22]研究指出,患者合并使用丙戊酸和拉莫三嗪,拉莫三嗪的血药浓度是单用拉莫三嗪患者的2倍。此外,也有一些学者指出,丙戊酸与拉莫三嗪联用具有相加或协同的药效学作用,使拉莫三嗪血药浓度增高时,也可增强抗癫痫疗效[23]。因此,当合用丙戊酸时应减少拉莫三嗪剂量;其次,应告知患者如果有变态反应的早期表现,如发热、血管性水肿、淋巴结肿大等症状,应立即就诊;当出现皮疹时,应立即停用拉莫三嗪,除非排除皮疹与药物相关。
2.1.4 卡马西平等肝药酶诱导剂 卡马西平是一种较强的肝药酶诱导剂,在加速自身代谢的同时,还可致丙戊酸代谢加速,半衰期缩短,血药浓度降低[24]。此外,丙戊酸与苯妥英钠(phenytoin)、苯巴比妥(phenobarbital)合并使用时,因为苯妥英钠、苯巴比妥的肝药酶诱导作用,也可使丙戊酸浓度显著下降[25-26]。因此与卡马西平、苯妥英钠、苯巴比妥合用时,需密切监测血药浓度以决定是否调整剂量,避免因血药浓度低于有效治疗浓度而导致癫痫发作[27]。
2.1.5 口服短效避孕药(oral contraceptives,OCs) 研究表明,OCs对丙戊酸产生影响主要通过诱导UGT 的表达[28],而UGT是丙戊酸体内重要的Ⅱ相代谢酶,介导约50%丙戊酸代谢,因此丙戊酸联合OCs使用时,可使患者丙戊酸清除率增加。GALIMBERTI等[29]研究发现,联合OCs时,丙戊酸体内总清除率增加21.5%,游离丙戊酸清除率增加45.2%。HERZOG等[30]研究结果显示,单用丙戊酸组血药浓度比联用OCs组高23.4%。研究提示,OCs可增加体内丙戊酸清除率,使癫痫发作风险增高。因此,在合并使用OCs时,应适量增加丙戊酸的给药剂量,并进行血药浓度监测。
遗传因素对丙戊酸的代谢有显著影响。丙戊酸3种代谢途径中,UGT、CYP代谢超过60%丙戊酸,其基因多态性位点可能通过改变酶活性,进而导致药物反应的个体差异。目前,关于丙戊酸的多态性研究也主要集中在CYP和UGT上,见
表1 基因多态性对丙戊酸代谢的影响
Tab.1 Influence of gene polymorphism on valproic acid metabolism
2.2.1 CYP酶基因多态性 研究表明,CYP是许多抗癫痫药物(antiepileptic drug,AEDs)的代谢酶,其多态性可影响AEDs的血药浓度[34]。体内约10%丙戊酸经由CYP2A6、CYP2B6、CYP2C9、CYP2C19等代谢,其中在CYP2A6、CYP2B6、CYP2C9代谢下生成4-ene-VPA等肝脏毒性代谢产物,这可能也是丙戊酸产生肝毒性等不良反应差异的原因之一[9,35]。同时考虑到上述基因多态性在中国人群中突变频率较高,因此CYP酶对丙戊酸血药浓度的影响,仍然值得临床注意。
孙妍萍等[36]在一项样本总数为98例的回顾性研究中发现CYP2A6*4突变基因型患者(CYP2A6*1/* 4 或* 4/* 4)的标准化血药浓度(浓度剂量比C/D)显著高于野生型患者CYP2A6*1/*1。此结果与文献[7,37]报道结果一致。欧江荣等[38]研究发现CYP2B6变异型组(CYP2B6*1/*6或CYP2B6* 6/*6)的平均血药浓度显著高于野生型组(CYP2B6*1、*1)。此结果与文献[7,39]报道研究结果相符。王育琴等[40]研究发现CYP2C19突变基因型(CYP2C19*1/*2,CYP2C19*1/*3,CYP2C19*2/*2,CYP2C19*3/*3)患者丙戊酸标准化血药浓度显著高于CYP2C19野生基因型(CYP2C19*1/*1)患者。此结果在另一项包含99例癫痫患者的研究中得以重现[41]。TAN等[7]在一项包含179 例中国癫痫患者的研究中发现,CYP2C9*3突变杂合子(CYP2C9*1/*3)标准化丙戊酸血药浓度显著高于CYP2C9*3 野生型患者(CYP2C9*1/*1)。上述两项结果与廖清船等[42]研究结果一致,131例服用丙戊酸单药治疗的癫痫患儿中,CYP2C9和CYP2C19基因多态性会影响丙戊酸血药浓度,突变型携带者标准化丙戊酸血药浓度显著高于野生型患者。
2.2.2 UGT酶基因多态性 UGT是人体内最主要的Ⅱ相结合反应的催化酶,它广泛分布于人体的肝、肾、肠等组织,代谢大量的外源性毒性物质和内源性物质,其基因多态性是个体间葡萄糖醛酸化活性差异的重要原因之一[43]。研究发现,UGT分为2个家族:UGT1、UGT2,3个亚家族:UGT1A、UGT2A、UGT2B,其中UGT1A、UGT2B是参与丙戊酸葡萄糖醛酸化的两种重要酶系,如UGT1A3、UGT1A6、UGT1A9,UGT2B7等基因对VPA体内代谢过程造成较大影响。此外,相对于CYP基因多态性对丙戊酸血药浓度的影响,UGT基因多态性对丙戊酸血药浓度的影响可能更大(后者代谢接近50%丙戊酸),更加值得临床关注[44]。
UGT2B7基因全长为16 kb,共5个内含子、6个外显子,编码529个氨基酸分子。研究显示,UGT2B7是VPA-G水解过程中活性最高的酶[45-48],因此UGT2B7基因多态性对丙戊酸的临床应用有重要影响。SUN等[49]纳入102例中国癫痫患者的研究中,分析发现UGT2B7 802C>T,含突变等位基因T的患者(CT型、TT型)的标准化血药浓度显著低于CC型患者,而UGT2B7 211GT(rs12233719)基因多态性对丙戊酸血药浓度无显著影响,但UGT2B7 211G>T仅在亚洲人群中发生突变,突变发生率韩国人12%[50],日本人18.5%[51],TAKEKUMA等[52]研究发现相对于野生型,突变型可使UGT代谢活性降低,因此关于UGT2B7 211G>T对丙戊酸血药浓度的影响,有待于前瞻性或大样本研究。此外,马虹英等[53]研究发现,UGT2B7 A268G基因多态性参与体内丙戊酸代谢,并影响其血药浓度,野生型患者丙戊酸血药浓度显著高于突变纯合子型患者。
UGT1A6是另一重要的Ⅱ相代谢酶之一,也是参与丙戊酸Ⅱ相代谢的主要代谢酶[57]。HUNG等[54]在一项162例单用丙戊酸治疗癫痫患者研究中发现,UGT1A6 19T>G(rs6759892),541A>G(rs2070959)和552A>C(rs1105879)等位基因携带者标准化血药浓度显著低于野生型,突变型患者需给予更高剂量。GUO等[55]研究也证实,在98例中国汉族癫痫患者中,UGT1A6 19T>G(rs6759892),541A>G(rs2070959)和552A>C(rs1105879)位点多态性对癫痫患者丙戊酸标准化血药浓度有显著性影响,突变纯合子标准化血药浓度显著低于野生型和突变杂合子。同时为排除各组间的年龄差异,GUO等[55]进一步对样本进行分层分析,0~4岁癫痫患儿与上述研究结果一致。但也有部分学者研究显示,UGT1A6 541A>G[56,58-59]、552 A>C[56,59]基因多态性与丙戊酸血药浓度无显著相关性。因此,通过进一步扩大样本研究和当前数据的Meta分析,将有助于阐明UGT1A6基因多态性的功能影响[60]。
此外,CHU等[56]研究表明,UGT1A3*5(A17G-T31C-G81A-A477G)突变基因型(包含突变纯合子和杂合子)标准化血药浓度显著低于野生型,提示UGT1A3*5可影响丙戊酸代谢,突变型患者必要时应注意增加剂量。
PPK是药动学的群体研究方法,它将群体统计模型与经典的药动学研究模型结合起来[61-63],从生物学和临床角度考虑药动学的实际问题,通常分析患者而不是健康受试者的病理生理因素,定量考察患者群体中药物浓度的决定因素,因此有助于制定和优化个体化给药方案。相对于经典药动学,PPK对临床稀疏数据和药动学参数变异的分析能力等方面具有明显的优势,已逐渐发展成为药动学研究与应用领域的主流方向之一,美国食品药品管理局(FDA)于1999年在新药开发指南中要求新药申请时需提供PPK参数[64-65]。近年来,PPK的应用范围越来越广,国内外已有借助PPK研究方法,定量探讨影响丙戊酸药动学参数的因素,预测丙戊酸体内血药浓度,进而实现丙戊酸的个体化给药。
丙戊酸是临床一线广谱抗癫痫药物,包含糖浆、缓释片等多种剂型,更适合儿童服用。因此,丙戊酸常作为癫痫患儿发作的首选AEDs,因此关于儿童丙戊酸的PPK研究报道也较多。此外,儿童与成人代谢酶活性也存在一定差异,有必要进行癫痫患儿的丙戊酸群体药动学研究,如介导丙戊酸代谢的重要Ⅱ相代谢酶UGT1A6、UGT1A9,其活性分别在患儿14个月[66]与2岁[67]时达到成人水平,UGT2B7在2~6个月时方可达到成人水平[68-69]。JIANG等[70]收集417例患者共计834例次稳态血药谷浓度,采用一级吸收和一房室模型,建立最终模型为
仅包含成人样本的VPA的PPK研究不多,多数研究未限制年龄,一般纳入儿童、成人共同研究。BLANCO-SERRANO等[74]回顾性收集208例共计534例日常TDM监测数据,运用NONMEM软件,分析丙戊酸日剂量、性别、年龄、总体质量、联用AEDs,建立最终模型:
目前,将基因多态性作为协变量影响因素纳入PPK模型的研究还较少。JIANG等[79]在一项包含287例中国癫痫患者的研究中发现,CYP2C19*2、 CYP2C9*3对VPA有显著影响,并首次将其上述两个基因纳入PPK模型中:
目前,关于UGT基因多态性对丙戊酸血药浓度影响的研究,有待进一步深入研究。同时,目前未见将UGT基因多态性作为协变量纳入PPK模型的研究[81]。
丙戊酸的药动学参数受年龄、体质量、联合用药、遗传基因等多种因素的影响,且其治疗窗窄,个体差异大,综合研究这些因素的影响,实现个体化给药治疗,是临床亟待解决的问题。PPK是将群体统计模型与经典的药动学原理结合起来,从临床实际及患者群体考虑问题,有助于制定和优化个体化给药方案。目前,基因多态性对丙戊酸药动学的影响还待进一步深入研究,将丙戊酸代谢酶基因多态性与PPK模型结合的研究也还很少,可考虑将基因多态性作为协变量纳入PPK 模型,定量评估丙戊酸的群体药动学参数,个体间和个体内变异,并对基因多态性对丙戊酸药动学的影响及影响程度进行量化,进而实现丙戊酸的个体化给药。
The authors have declared that no competing interests exist.
| [1] |
Abstract The anticonvulsant properties of VPA (valproic acid), a branched short-chain fatty acid, were serendipitously discovered in 1963. Since then, therapeutic roles of VPA have increased to include bipolar disorder and migraine prophylaxis, and have more recently been proposed in cancer, Alzheimer's disease and HIV treatment. These numerous therapeutic roles elevate VPA to near 'panacea' level. Surprisingly, the mechanisms of action of VPA in the treatment of many of these disorders remain unclear, although it has been shown to alter a wide variety of signalling pathways and a small number of direct targets. To analyse the mechanism of action of VPA, a number of studies have defined the structural characteristics of VPA-related compounds giving rise to distinct therapeutic and cellular effects, including adverse effects such as teratogenicity and hepatotoxicity. These studies raise the possibility of identifying target-specific novel compounds, providing better therapeutic action or reduced side effects. This short review will describe potential therapeutic pathways targeted by VPA, and highlight studies showing structural constraints necessary for these effects.
[本文引用:1]
|
| [2] |
[Article in French]
[本文引用:1]
|
| [3] |
1. The kinetic profile of sodium valproate (VPA) and the GABA levels were studied in discrete brain areas of the rat after an i.p. injection of 200 mg/kg. The results were discussed comparatively with GABA-T and GAD activities reported in the literature. 2. VPA was rapidly distributed in brain areas; its concentrations, its kinetic parameters and the GABA levels after the drug administration were not uniform in the different brain areas studied. 3. The results showed a particular relation of the VPA to the olfactory bulbs; in this specific area the VPA effect on GABA level was stronger; the VPA apparent half life of elimination was longest; the VPA apparent disappearance rate constant was smallest; the initial GABA level was higher; the activities of GABA-T and GAD were higher than in other brain areas studied except the hypothalamus. 4. These data were correlated with the role of the olfactory bulbs in the behaviour of the rodents.
[本文引用:1]
|
| [4] |
|
| [5] |
ABSTRACT Therapeutic drug monitoring (TDM) of antiepileptic drugs (AEDs) has made it possible to study the individual variations in drug utilization, to reveal noncompliance in patients and for quality assurance aspects. Even if there is a shortage of data from randomized controlled studies concerning the effectiveness of using TDM as an aid to dosage adjustment, experience from nonrandomized investigations and long-lasting clinical experience have shown that TDM of both older and newer AEDs may be of clinical benefit if used appropriately. The main situations for TDM include: after starting treatment to provide a baseline steady-state concentration for further evaluation of an individual therapeutic concentration; after change in drug dosage, in particular when nonlinear kinetics apply; at therapeutic failure to sort out a pharmacokinetic explanation for uncontrolled seizures or side effects; in case of drug interactions; and when pharmacokinetic changes due to physiological or pathological changes are foreseen (e.g., age-dependent conditions [children, elderly], pregnancy, hepatic disease, renal disease or gastrointestinal conditions potentially affecting drug absorption) and change in drug formulation (brand name/generic). Recently, new terminology and definitions have been suggested by the International League Against Epilepsy. The reference range is a range of drug concentrations quoted by laboratories and is not a therapeutic range. Emphasis should be placed on the concept of an individual therapeutic concentration.
[本文引用:1]
|
| [6] |
Five distinct acyl-CoA dehydrogenases are currently known. These are short, medium, long and 2-methyl-branched-chain acyl-CoA dehydrogenases, and isovaleryl-CoA dehydrogenase. We tested these five acyl-CoA dehydrogenases for their ability to dehydrogenase valproyl-CoA using pure enzyme preparations isolated from rat liver mitochondria. The activities of the pure human short-chain, medium-chain, and isovaleryl enzymes purified from post-mortem livers, and a long-chain acyl-CoA dehydrogenase preparation partialy purified from placental mitochondria, were also tested. Valproyl-CoA was dehydrogenated at a significant rate (0.167 渭mol/min per mg protein) only by rat 2-methyl-branched-chain acyl-CoA dehydrogenase. Human 2-methyl-branched-chain acyl-CoA dehydrogenase has not been purified; therefore, it could not be tested. Since four other human acyl-CoA dehydrogenases did not dehydrogenate isobutyryl-CoA, 2-methylbutyryl-CoA (obligatory intermediates from valine and isoleucine, respectively) nor valproyl-CoA, it is reasonable to assume that valproyl-CoA is dehydrogenated by 2-methyl-branch-chain acyl-CoA dehydrogenase in man as well. We identified 2-propyl-2-pentenoyl-CoA as the reaction product from valproyl-CoA by mass spectral analysis of the acyl moiety. Valproyl-CoA, at 0.3 mM, moderately inhibited human acyl-CoA dehydrogenases with the exception of the long-chain enzyme. 5 mM free valproic acid inhibited the activities of various acyl-CoA dehydrogenases only very weakly.
[本文引用:1]
|
| [7] |
To investigate influences of the functional polymorphisms of Cytochrome P450 isozymes 2A6 (CYP2A6), 2B6 (CYP2B6), and 2C9 (CYP2C9) on pharmacokinetics of VPA in vivo. In the study, we analyzed the genotypes of CYP2A6, CYP2B6, and CYP2C9 and their contribution to the steady-state standardized plasma VPA concentrations in 179 subjects with epilepsy of a Northern Han Chinese population. The genotypes were detected by the polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP). The subjects with one or two variant CYP2A6*4 alleles showed higher mean plasma VPA concentrations compared with non-*4 alleles [(3.4 ± 0.4) μg kg ml 611 mg 611 vs. (3.6 ± 0.4) μg kg ml 611 mg 611, p = 0.0055]. A significant difference [one-way ANOVA ( p = 0.0203)] was also found between mean plasma VPA concentrations and the CYP2B6 genotypes. In addition, subjects with the heterozygous genotype CYP2C9*3 had higher mean plasma VPA concentrations than did those subjects with the wild-type genotype [(3.9 ± 0.4) μg kg ml 611 mg 611 vs. (3.4 ± 0.4) μg kg ml 611 mg 611, p = 0.0001]. The presently evaluated variant alleles in the CYP2A6, CYP2B6, and CYP2C9 genes may explain part of the substantial variability in VPA pharmacokinetics between different subjects.
[本文引用:2]
|
| [8] |
丙戊酸是临床常用一线抗癫痫药,对癫痫小发作、大发作和肌阵挛发作疗效显著.丙戊酸的给药剂量与血药浓度 的相关性差,临床药物安全治疗浓度范围较窄.肝毒性是丙戊酸较严重的毒副作用,可诱发肝脏脂质病变甚至是致死性肝坏死,在一定程度上限制了丙戊酸的临床应 用.大量研究表明,丙戊酸的肝毒性与其代谢特征息息相关.本文综述了丙戊酸的代谢动力学及其肝毒性的相关性,以期为丙戊酸的临床安全用药提供一定的科学依 据.
[本文引用:1]
|
| [9] |
<p>丙戊酸钠为常用广普抗癫痫药物,在临床上广泛应用于治疗神经性疾病,但是其可引起严重的肝毒性。尤其2岁以下儿童发生致死性肝毒性的风险显著增加,严重时甚至发生致死性、急性肝坏死。2-丙基-4-戊烯酸(2-propyl-4-pentanoic acid, 4-ene-VPA)为丙戊酸在体内经细胞色素P450(cytochrome P450,CYP450)代谢产生的代谢产物,研究证实其和丙戊酸导致的严重肝毒性有着密切的关系。本文就丙戊酸代谢产物4-ene-VPA近年的研究进展做一综述,为临床安全用药提供参考依据。</p>
[本文引用:2]
|
| [10] |
Valproic acid (VPA) is a widely used anticonvulsant that is also approved for mood disorders, bipolar depression, and migraine. In vivo, valproate is metabolized oxidatively by cytochromes P450 and 尾-oxidation, as well as conjugatively via glucuronidation. The acyl glucuronide conjugate (valproate-glucuronide or VPAG) is the major urinary metabolite (30鈥50% of the dose). It has been hypothesized that glucuronidation of antiepileptic drugs is spared over age, despite a known decrease in liver mass. The formation rates of VPAG in a bank of elderly (65 years onward) human liver microsomes (HLMs) were measured by liquid chromatography/tandem mass spectrometry and compared with those in a younger (2鈥56 years) HLM bank. In vitro kinetic studies with recombinant UDP-glucuronosyltransferases (UGTs) were completed. A 5- to 8-fold variation for the formation of VPAG was observed within the microsomal bank obtained from elderly and younger donors. VPAG formation ranged from 6.0 to 53.4 nmol/min/mg protein at 1 mM substrate concentration (n = 36). The average velocities at 0.25, 0.5, and 1 mM VPA were 7.0, 13.4, and 25.4 nmol/min/mg protein, respectively, in the elderly HLM bank. Rates of VPAG formation were not significantly different in the HLM bank obtained from younger subjects. Intrinsic clearances (Vmax/Km) for several cloned, expressed UGTs were determined. UGT1A4, UGT1A8, and UGT1A10 also were found to catalyze the formation of VPAG in vitro. This is the first reported activity of these UGTs toward VPA glucuronidation. UGT2B7 had the highest intrinsic clearance, whereas UGT1A1 demonstrated no activity. In conclusion, our investigation revealed no differences in VPAG formation in younger versus elderly HMLs and revealed three other UGTs that form VPAG in vitro.
[本文引用:1]
|
| [11] |
Valproic acid (VPA) is a widely used anticonvulsant that is also approved for mood disorders, bipolar depression, and migraine. In vivo, valproate is metabolized oxidatively by cytochromes P450 and 尾-oxidation, as well as conjugatively via glucuronidation. The acyl glucuronide conjugate (valproate-glucuronide or VPAG) is the major urinary metabolite (30鈥50% of the dose). It has been hypothesized that glucuronidation of antiepileptic drugs is spared over age, despite a known decrease in liver mass. The formation rates of VPAG in a bank of elderly (65 years onward) human liver microsomes (HLMs) were measured by liquid chromatography/tandem mass spectrometry and compared with those in a younger (2鈥56 years) HLM bank. In vitro kinetic studies with recombinant UDP-glucuronosyltransferases (UGTs) were completed. A 5- to 8-fold variation for the formation of VPAG was observed within the microsomal bank obtained from elderly and younger donors. VPAG formation ranged from 6.0 to 53.4 nmol/min/mg protein at 1 mM substrate concentration (n = 36). The average velocities at 0.25, 0.5, and 1 mM VPA were 7.0, 13.4, and 25.4 nmol/min/mg protein, respectively, in the elderly HLM bank. Rates of VPAG formation were not significantly different in the HLM bank obtained from younger subjects. Intrinsic clearances (Vmax/Km) for several cloned, expressed UGTs were determined. UGT1A4, UGT1A8, and UGT1A10 also were found to catalyze the formation of VPAG in vitro. This is the first reported activity of these UGTs toward VPA glucuronidation. UGT2B7 had the highest intrinsic clearance, whereas UGT1A1 demonstrated no activity. In conclusion, our investigation revealed no differences in VPAG formation in younger versus elderly HMLs and revealed three other UGTs that form VPAG in vitro.
[本文引用:1]
|
| [12] |
The effects of salicylic acid on the pharmacokinetics of valproic acid were investigated in bile-exteriorized rats. A 50 mg/kg bolus dose of sodium valproate was injected iv to Long Evans rats with and without (control) prior treatment by constant infusion of salicylate to keep it at steady state plasma level (about 250 micrograms/ml). The plasma elimination of valproic acid followed a monoexponential decline in both salicylate-treated and control rats. A significant increase (p less than 0.01) in the disposition rate constant (kel), the volume of distribution (Vd), and the total clearance (Cltot) as well as a significant decrease (p less than 0.01) in the AUC and the elimination half-life (t1/2) were observed in the salicylate-treated rats. In spite of the significantly lowered total plasma level and increased unbound fraction of valproic acid in the salicylate-treated rats, there were no significant differences in unbound valproic plasma levels and unbound valproate pharmacokinetic parameters. The biliary excretion of unchanged and conjugated valproate was not significantly different between the two groups. The in vitro plasma-unbound fractions (fu) of valproic acid were significantly increased (p less than 0.01) in the presence of salicylic acid. The apparent dissociation constant of plasma protein binding for valproic acid was increased from 0.287 to 1.204 mM in the presence of salicylic acid. These findings indicate that the pharmacokinetic changes of valproic acid in the presence of salicylic acid were consistent with the elevation in the plasma-unbound fraction of valproic acid due to displacement from plasma protein-binding sites by salicylic acid.(ABSTRACT TRUNCATED AT 250 WORDS)
[本文引用:2]
|
| [13] |
In five of six epileptic children who were taking 18 to 49 mg/kg/day valproic acid (VPA), the steady-state serum free fractions of VPA rose from 12% to 43% when antipyretic doses of aspirin were also taken. Mean total VPA half-life (t1/2) rose from 10.4 +/- 2.7 to 12.9 +/- 1.8 hr and mean free VPA t1/2 rose from 6.7 +/- to 2.1 to 8.9 +2- 3.0 hr when salicylate was present in the serum. The in vitro albumin binding association constant (ka) for VPA was decreased by salicylate, but the in vivo ka value was not affected. The 12-hr (trough) concentrations of both free and total VPA were higher in the presence of serum salicylate in five of six patients. Renal excretion of unchanged VPA decreased in five of six patients, but the VPA carboxyl conjugate metabolite-excretion patterns were not consistently affected. Salicylate appeared to displace VPA from serum albumin in vivo, but the increased VPA t1/2 and changes in VPA elimination patterns suggest that serum salicylate also altered VPA metabolism.
[本文引用:1]
|
| [14] |
Abstract Plasma levels of valproic acid (VPA) are decreased by concomitant use with carbapenem antibiotics, such as panipenem (PAPM). One of the plausible mechanisms of this interaction is the inhibition of VPA glucuronide (VPA-G) hydrolysis by carbapenems in the liver. To elucidate this interaction mechanism, we purified VPA-G hydrolase from human liver cytosol, in which the hydrolytic activity was mainly located. After chromatographic purification, the VPA-G hydrolase was identified as acylpeptide hydrolase (APEH). APEH-depleted cytosol, prepared by an immunodepletion method, completely lacked the hydrolytic activity. These results demonstrate that APEH is a single enzyme involved in PAPM-sensitive VPA-G hydrolysis in cytosol. In addition, the hydrolytic activity of recombinant human APEH was inhibited by PAPM and the inhibition profile by typical esterase inhibitors (diisopropyl fluorophosphate, 5,5'-dithiobis(2-nitrobenzoic acid), p-chloromercuribenzoic acid, and d-saccharic acid 1,4-lactone) was similar to that of human liver cytosol. Cytosolic VPA-G hydrolase activity was slightly inhibited by cholinesterase and carboxylesterase inhibitors. beta-Glucuronidase activity remained in APEH-depleted cytosol, whereas VPA-G hydrolase activity was completely abolished. Thus, either cholinesterase, carboxylesterase, or beta-glucuronidase in cytosol would not be involved in VPA-G hydrolysis. Taken together, APEH plays a major role in the PAPM-sensitive VPA-G hydrolysis in the liver. These findings suggest that APEH could be a key enzyme for the drug interaction of VPA with carbapenems via VPA-G hydrolysis.
[本文引用:1]
|
| [15] |
Abstract We have reported that inhibition of acylpeptide hydrolase (APEH), identified as valproic acid glucuronide hydrolase in human liver cytosol, by carbapenem antibiotics could lead to a decrease of plasma levels of valproic acid. In this study, we examined the inhibition mechanism using human liver cytosol and purified porcine APEH with a similar property to human counterpart. After preincubation of human liver cytosol with panipenem or meropenem for 3065min, the inhibition of APEH activity was 20-fold stronger than that without preincubation. Porcine APEH activity inhibited by meropenem did not recover after dialysis. Meropenem bound to porcine APEH and the binding was blocked by a serine hydrolase inhibitor, diisopropyl fluorophosphate. Open β-lactam ring form of meropenem did not affect APEH activity in human liver cytosol. Likewise, other antibiotics, which have a different heterocycle adjacent to the β-lactam ring with an opposite configuration of the side chain from carbapenems, did not inhibit APEH activity. In conclusion, carbapenems inhibit APEH in both reversible and true irreversible manner and the irreversible inhibition is partially explained by binding to the active serine of APEH. The closed β-lactam ring is essential for inhibition and the heterocycle and/or the configuration of side chain would be important.
[本文引用:1]
|
| [16] |
Case We describe here a rare case in which valproic acid (VPA) levels were affected by ertapenem but not by meropenem even though ertapenem and meropenem are in the same carbapenem class. A 68-year-old Filipino male treated with valproate for epilepsy and ertapenem for an infectious disease had decreased VPA levels during the first day of ertapenem therapy. His VPA level increased soon after terminating ertapenem therapy. Two types of carbapenems had different drug reactions with concomitant use of VPA in this patient. Conclusions Closer monitoring of VPA concentrations are necessitated using carbapenems for treating infection in patients being administered VPA. Another option is the use of anti-epileptic drugs other than VPA if concomitant use with a carbapenem is warranted.
[本文引用:1]
|
| [17] |
The plasma concentrations of valproic acid (VPA) are known to decrease during the concomitant administration of carbapenem antibiotics, such as meropenem, imipenem, and ertapenem. This study summarizes 6 cases of drug-drug interactions between VPA and carbapenem antibiotics.To investigate the onset and severity of the reductions in the concentration of VPA in patients with or without the coadministration of carbapenem antibiotics, the authors performed a retrospective evaluation of therapeutic drug monitoring (TDM) reports that described a decrease in the serum concentrations of VPA during the concomitant use of carbapenem antibiotics from January 2008 to December 2010 in the Seoul National University Hospital. The evaluated TDM reports included 6 cases. The decrement ratio of the VPA serum concentration was calculated from the TDM reports, and the change in the half-life of the VPA was also estimated.Six cases presented with changes in the VPA serum concentration before and after the administration of carbapenem antibiotics. (Three cases were treated with meropenem, 2 were treated with ertapenem, and 1 was treated with imipenem.) The VPA concentrations reduced by (mean 卤 SD) 88.7 卤 5.3% (3 cases of meropenem), 74.0 卤 9.8% (2 cases of ertapenem), and 73.3% (1 case of imipenem), respectively, and the half-life of VPA reduced by 80.1 卤 9.0%, 64.4 卤 24.2%, and 50.6%, respectively.The interaction between VPA and carbapenem antibiotics caused decreases in the VPA serum concentrations; the extent of this decrease was greater in the meropenem-treated patients than in the imipenem-treated or ertapenem-treated cases. Because the therapeutic effect of VPA depends on its serum concentration, it should be recognized that there may be a loss of seizure control in patients using VPA with carbapenem antibiotics.
[本文引用:1]
|
| [18] |
SummaryWhat is known and objectiveA series of studies have indicated that valproic acid (VPA) plasma concentration decreased rapidly when used concomitantly with carbapenem antibiotics, inc ...
[本文引用:2]
|
| [19] |
Valproic acid (VPA) is the most commonly used antiepileptic drug in pediatric patients, but its major drawback is its multiple pharmacological interactions.To study children who had been simultaneously treated with carbapenems and valproic acid, considering drug levels, pharmacological interactions and clinical follow-up.Retrospective study of children who simultaneously received treatment with VPA and carbapenems between January 2003 and December 2011. Demographic variables, indication of treatment, dose, VPA plasma levels, interactions, clinical manifestations and medical management were analyzed.28 children with concomitant treatment with both drugs were included in the study. 64.3% were males. 78.6% of the interactions were observed in the Intensive Care Unit. 60.7% of children had been previously treated VPA and its major indication were generalized seizures. Basal plasma levels of VPA were recorded in 53% and at 24hafter admittance in 60%. "40% of basal VPA levels were below therapeutic range prior to the administration of carbapenem. After the introduction of carbapenem 88% of level determinations were below therapeutic range". 54.5% of the patients that were chronically receiving VPA and had good control of epilepsy before admission had seizures during the coadministration. One patient that was on VPA before admission but with bad control of epilepsy worsened, and one patient that acutely received VPA did not achieve seizure freedom. In these cases it was necessary to either increase VPA dose or change to a different antiepileptic drug.Little is known about the mechanism of pharmacologic interactions between carbapenems and VPA, but it leads to a reduction in plasma levels that may cause a loss of seizure control, so simultaneous use of both drugs should be avoided when possible. If not, VPA levels should be monitored.
[本文引用:1]
|
| [20] |
Lamotrigine and valproic acid are well-tolerated anticonvulsants, but frequently associated with severe cutaneous reactions, such as the Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis, when used in combination. We report a case of SJS likely induced by the use of a lamotrigine and valproic acid regimen and as a dental surgeon it is important to identify such lesion and report to pharmacovigilance.
[本文引用:1]
|
| [21] |
Stevens-Johnson Syndrome (SJS) and toxic epidermal necrolysis (TEN) are diseases within the spectrum of severe cutaneous adverse reactions affecting skin and mucous membranes. Antiepileptic drugs (AEDs) are used in combination, leading to potential pharmacokinetic or pharmacodynamic interactions, causing more adverse effects than might occur when the AED is taken as monotherapy. Here, we report a rare case of SJS triggered by a combination of clobazam, lamotrigine and valproic acid in a 7-year-old boy. Because of inadequate seizure control, lorazepam was replaced with clobazam. Four weeks after the addition of clobazam, the patient developed SJS with a generalized rash, fever, with liver and kidney involvement, and eosinophilia one week after the initiation of treatment. All antiepileptic drugs were discontinued, and intravenous methylprednisolone, prophylactic systemic antibiotics, intravenous fluid supplement, antipyretic, special wound care, and supportive medical care for SJS were administered. He was discharged in a stable condition on the 18th day. Our case suggests that a drug-drug interaction between valproate, lamotrigine and clobazam contributed to the development of SJS. When the clobazam was added to valproic acid and lamotrigine co-medication, the lamotrigine dose should have been decreased.
[本文引用:1]
|
| [22] |
Lamotrigine (LTG) is a new antiepileptic drug (AED), chemically unrelated to the drugs in current use. Previous studies have shown that LTG has only a limited effect on other AEDs, but its own metabolism can be strongly induced or inhibited by the comedication. We investigated the influences of carbamazepine (CBZ), phenytoin (PHT), phenobarbital (PB), valproic acid (VPA), and combinations of these drugs on the serum concentration of LTG. A total of 588 blood samples from 302 patients were analyzed. The mean duration of LTG therapy was 141 +/- 137 days (mean +/- SD). A patient was only considered twice in this study if his or her comedication had been changed. The LTG serum concentration in relation to LTG dose/body weight (level-to-dose ratio, LDR, microgram/ml/mg/kg) was calculated and compared for different drug combinations. The results showed that comedication had a highly significant (p 0.05, multiple comparisons). The mean LTG concentrations in patients on comedication with VPA were about two times higher than on patients on LTG monotherapy or on comedication without VPA (5.0 vs. 2.6 micrograms/ml), despite the LTG doses being half as high (3.0 vs. 5.9 mg/kg). The correlations of the serum concentrations and doses of CBZ, PB, PHT, and VPA with the LDR of LTG were only weak or not significant. Furthermore, the distribution of LTG serum concentrations and dosages was compared with the tentative therapeutic range for the LTG concentration (1-4 micrograms/ml), proposed by some investigators, and the recommendations for the LTG dosage. Remarkable discrepancies were observed. The comedication has an important influence on the LTG concentration and should be considered in LTG dosage.
[本文引用:1]
|
| [23] |
目的考察丙戊酸(VPA)联合 拉莫三嗪(LTG)治疗时,癫痫患者体内丙戊酸对拉莫三嗪药代动力学的影响,以及联合用药的有效性及安全性。方法共收集符合入选标准的癫痫患者病例294 例,拉莫三嗪+丙戊酸合用组(A)72例,拉莫三嗪单用组(B)92例,丙戊酸单用组(C)130例,分析各组之间血药浓度、临床疗效、不良反应的差异。 结果 A组的拉莫三嗪血药浓度与标准化血药浓度(CDR_(拉莫三嗪))分别是B组2.23、1.93倍(P0.01,P0.01),A组与C组的丙戊酸浓度和 CDR_(丙戊酸)相比,差异无统计学意义(P0.05,P0.05)。A组中不同丙戊酸浓度对拉莫三嗪浓度及CDR_(拉莫三嗪)无影响 (P0.05)。A组的控制率显著高于B组和C组(P0.05,P0.05)。3组不良反应发生率差异均无统计学意义(P0.05)。结论拉莫三嗪与丙戊 酸联合治疗时,临床疗效显著,但丙戊酸可增加拉莫三嗪的血药浓度,在合用时需调整拉莫三嗪剂量,以减少不良反应的发生。
[本文引用:1]
|
| [24] |
Objective: The aim of the present study was to build population pharmacokinetic models for the clearance of valproate (VPA) in 2 separate populations of Serbian patients with epilepsy, children and adults. Methods: Analysis was performed using 65 and 63 steady-state concentrations of VPA collected from 58 children and 60 adult epileptic patients, respectively. Mean values for total body weight and age were 27.07 ± 13.08 kg and 7.21 ± 3.63 years in the pediatric population, and 69.67 ± 15.60 kg and 33.97 ± 16.41 years in the adult population. The one-compartment model with first order elimination and without absorption was used from the PREDPP (Prediction for Observation Population Pharmacokinetics) library of NONMEM software. Results: The derived final models show that VPA clearance increased with total body weight of patients in both populations. However, the carbamazepine comedication was the main determinant of the final model in children whereas phenobarbitone comedication was the most important factor in the adult population. The magnitudes of these effects were +0.159 lh
[本文引用:1]
|
| [25] |
|
| [26] |
Abstract INTRODUCTION: There are no published reports on pediatric phenytoin toxicity, resulting from the drug interaction between phenytoin and valproic acid. CASE DESCRIPTION: A 12-year-old patient with refractory epilepsy syndrome presented with phenytoin toxicity, following a concomitant treatment with phenytoin, valproic acid, and lamotrigine. The phenytoin concentration detected in the capsules used by the patient was in accordance with the prescribed dose and was appropriate for the age and weight of the patient. However, a supratherapeutic phenytoin serum concentration was observed (21.92 脗碌g phenytoin/mL of blood). Consequently, the phenytoin dose was reduced, and the patient was monitored; 24 hours later the patient did not present with any signs/symptoms of toxicity. DISCUSSION: Despite the appropriate phenytoin concentration in the capsules, the patient presented with phenytoin toxicity. This toxicity likely resulted from the drug interaction between phenytoin and valproic acid that leads to phenytoin displacement from plasmatic proteins and inhibits phenytoin metabolism, thereby increasing the concentration of free drug in the serum.
[本文引用:1]
|
| [27] |
|
| [28] |
ABSTRACT Uridine diphosphate glucuronosyltransferases (UGTs) catalyze the glucuronidation of a wide range of xenobiotics and endogenous substrates. However, there is a lack of information concerning the response of human UGTs to inducers, and this observation prompted the current investigation. The glucuronidation of estradiol (3- and 17-positions), naphthol, propofol, and morphine (3- and 6-positions) was assessed against a battery of recombinant human UGTs to determine selective glucuronidation reactions for induction studies. The potential induction of the glucuronidation of estradiol at the 3-position, naphthol, propofol, and morphine at the 3-position was subsequently investigated in cultured primary human hepatocytes against a range of prototypic inducers including dexamethasone, 3-methylcholanthrene (3-MC), phenobarbital, rifampicin, and omeprazole. Treatment with 3-MC induced estradiol-3-glucuronidation (up to 2.5-fold) in four of five donors investigated. Statistically significant increases in naphthol glucuronidation (up to 1.7-fold) were observed following treatment with carbamazepine. UGT1A9-mediated propofol glucuronidation was induced by phenobarbital (up to 2.2-fold) and rifampicin (up to 1.7-fold). However, treatment with alpha-naphthoflavone and tangeretin resulted in a decrease in propofol glucuronidation (30% of control values). Statistically significant induction of morphine-3-glucuronidation was observed in at least three donors following treatment with phenobarbital, rifampicin, and carbamazepine. Each UGT isoform investigated displayed a distinct induction profile. Although statistically significant increases in glucuronidation were observed for each reaction studied, the level of induction was less than that observed for CYP1A2 or CYP3A4 and exhibited a large interdonor variability. The clinical relevance of the induction responses obtained in this study is unclear.
[本文引用:1]
|
| [29] |
To determine potential changes in total and unbound serum valproic acid (VPA) concentrations at steady-state during a cycle of intake of combined hormonal contraceptive (HC) steroids.Blood samples were collected from nine women stabilized on VPA monotherapy on two separate randomized occasions: (i) at the end of the 4- to 7-day HC-free interval, and (ii) on the last day of the HC intake period. Trough concentrations of VPA in serum and serum ultrafiltrates were determined by fluorescence polarization immunoassay.In all women, total and unbound VPA concentrations were higher during the HC-free interval than during HC intake (means +/- SD: 425 +/- 184 vs. 350 +/- 145 micromol/L, respectively, for total VPA, p = 0.002, and 55 +/- 37 vs. 39 +/- 25 micromol/L, respectively, for unbound VPA, p = 0.005). Compared with the HC-free interval, HC intake was associated with a mean 21.5% increase in VPA total apparent oral clearance (from 8.0 +/- 5.2 to 9.7 +/- 6.4 ml/h/kg, p = 0.01) and a 45.2 % increase in VPA unbound apparent oral clearance (from 79 +/- 81 to 115 +/- 121 ml/h/kg, p = 0.029).The apparent oral clearance of total and unbound VPA increases during the HC intake period compared with the HC-free interval, probably due to induction of glucuronosyltransferase by ethinylestradiol. The magnitude of the change varies across individuals, being potentially clinically relevant in some cases. Serum VPA concentrations should be monitored when adding or discontinuing HC steroids, and possibly during the on-off intervals of a HC cycle.
[本文引用:1]
|
| [30] |
To determine whether 1) combined oral contraceptive (COC) use affects serum levels of valproate (VPA) as well as lamotrigine (LTG) and 2) the naturally occurring high (mid-luteal) and low (early-mid follicular) reproductive steroid level phases of the menstrual cycle might affect antiepileptic drug levels as well.This investigation compared serum antiepileptic drug levels at two timepoints during a single menstrual cycle in four groups of women with epilepsy: 12 on VPA, 12 on VPA plus COC (VPA-COC), 12 on LTG, and 12 on LTG plus COC (LTG-COC).Both VPA and LTG levels were lower (p < 0.01) on active COC than on inactive pill with median declines of 23.4% for the VPA-COC group and 32.6% for the LTG-COC group. Serum LTG levels showed a notable but not significant 31.3% median decline during the mid-luteal phase compared to the early-mid follicular phase in the non-COC group. The non-COC valproate group showed the least change of any group between the two measured timepoints with a decline of 8.3% (p = NS).The findings suggest that valproate (VPA), like lamotrigine (LTG), has substantially and significantly lower serum levels while women take active combined oral contraceptives as compared to inactive pills. Larger sample sizes will be required to determine whether LTG levels may drop significantly also during the luteal (high steroid) phase of natural menstrual cycles and whether VPA levels may show greater stability in levels across the phases of the menstrual cycle.
[本文引用:1]
|
| [31] |
|
| [32] |
Drug interactions in the neurosciences intensive care unit (NICU) may involve antiepileptic drugs and warfarin. Most commonly used antiepileptic drugs are either potent hepatic enzyme inducers or inhibitors and they affect the metabolism of warfarin. Valproic acid also displaces warfarin from the protein binding sites resulting in significant INR changes but this type of drug interaction is less well known.Case report.A 71 year-old female patient with a glioblastoma multiforme presented to the NICU with refractory partial complex seizures. Patient was on warfarin for a prior deep venous thrombosis. After treatment with levetiracetam, seizures recurred and intravenous loading with valproic acid was administered, but resulted in a rapid increase in international normalized ratio (INR) to 7.6. A MRI of the brain showed hemorrhagic tumor, but no new major bleeding.With both acidic drugs present, a loading dose of valproic acid may displace warfarin from the protein binding sites resulting in redistribution of warfarin in free active form and lead to a rapid increase in INR.
[本文引用:1]
|
| [33] |
Guthrie SK, Stoysich AM, Bader G, Hilleman DE.
[本文引用:1]
|
| [34] |
|
| [35] |
Cytochrome P450-dependent desaturation of the anticonvulsant drug valproic acid (VPA) results in formation of the hepatotoxin, 4-ene-VPA. Polytherapy with other anticonvulsants which are known P450 inducers increases the flux through this bioactivation pathway. The aim of the present study was to identify specific, inducible forms of human liver P450 which catalyze terminal desaturation of VPA. Oxidized VPA metabolites formed in an NADPH-dependent manner by human liver microsomes were quantified by gas-chromatography/mass spectrometry. In vitro reaction conditions were established which reflected the product profile found in vivo. Production of 4-ene-VPA by microsomal P450s could be inhibited significantly by coumarin, sulfaphenazole and diethyldithiocarbamate, but not by triacetyloleandomycin, quinidine or furafylline. Recombinant human CYP3A4 did not form detectable levels of 4-ene-VPA and, of nine additional isoforms expressed in either HepG2 or lymphoblastoid cells which were screened for VPA desaturase activity, only CYP2C9 and CYP2A6 formed detectable levels of metabolite. Consequently, CYP3A4, the isoform usually associated with induction by anticonvulsants cannot be responsible for the enhanced 4-ene-VPA formation that occurs during polytherapy. Instead, enhanced activity in vivo likely results from induction of CYP2A6 and/or CYP2C9.
[本文引用:1]
|
| [36] |
目的 探讨细胞色素P450 2A6(CYP2A6)基因多态性对丙戊酸钠血药浓度的影响.方法 选择单药服用丙戊酸钠的癫NFDA1患者98例,应用巢式PCR(nested-primer polymerase chain reaction)方法分析其CYP2A6基因型,分析等位基因CYP2A6*1及CYP2A6*4;同时应用荧光偏振免疫法(FPIA)测定患者丙戊酸 钠的血药浓度.结果 98例患者中CYP2A6基因型为*1/*1者73例(74.5%),*1/*4者24例(24.5%),*4/*4者1例(1.0%),根据基因型分为 A组(CYP2A6*1/*1)和B组(CYP2A6*1/*4或CYP2A6*4/*4).B组患者丙戊酸钠的标准血药浓度平均值 (4.1393±0.2793)较A组(3.3486±0.3919)高,差异有统计学意义(P<0.05). 结论 CYP2A6基因多态性影响丙戊酸钠的血药浓度,含有CYP2A6*4等位基因的患者应用丙戊酸钠应较常规降低用量.
[本文引用:1]
|
| [37] |
|
| [38] |
|
| [39] |
目的探讨细胞色素P450(CYP)2A6及CYP286等位基因多态性与丙戊酸钠血药浓度的关系。方法选择165例服用丙戊酸钠单药治疗且无肝肾功能异常的癫痫患者,应用多聚酶链反应(PCR)方法分别进行CYP2A6(95例)和CYP286(70例)等位基因多态性频率分析;应用荧光偏振免疫法(FPIA)测定含不同等位基因患者丙戊酸钠的血药浓度。结果95例患者中,CYP2A6*4等位基因频率为13.2%,CYP2A6*4等位基因携带者丙戊酸钠的血药浓度[(4.23±0.27)mg/ml]明显高于非CYP2A6*4等位基因携带者[(3.35±0.38)mg/ml](P〈0.05);70例患者中,CYP286*6等位基因频率为24.3%,CYP286*6等位基因携带者丙戊酸钠的血药浓度[(4.12±0.34)mg/ml]明显高于非CYP286*6等位基因携带者[(3.07±0.28)mg/ml](P〈0.05)。结论CYP2A6或(和)CYP286等位基因多态性均影响丙戊酸钠的血药浓度,CYP2A6*4或(和)CYP286*6等位基因携带者丙戊酸钠用量应低于常规剂量,以减少不良反应的发生和避免药物资源的浪费。
[本文引用:0]
|
| [40] |
目的:寻找丙戊酸药物浓度与CYP2C19基因多态性的关系,以便临床根据患者的基因型进行个体化给药。方法:运用血药浓度监测仪测定患者血药浓度和变性高效液相色谱法检测癫痫患者的CYP2C19基因多态性位点,对二者结果进行相关性分析。结果:51名汉族癫痫患者中有29名携带突变型CYP2C19基因,其中19名(65.52%)患者丙戊酸实际血药浓度较预期的血药浓度升高,血药浓度分布曲线右移。结论:CYP2C19参与丙戊酸的代谢。对于含突变型CYP2C19基因的患者应给予小剂量丙戊酸,以减少药物不良反应的发生和药物资源的浪费。
[本文引用:1]
|
| [41] |
|
| [42] |
目的 探讨细胞色素P450酶2A6(CYP2A6)、2B6(CYP2B6)、2C9(CYP2C9)和2C19(CYP2C 19)基因多态性对丙戊酸钠血药浓度的影响.方法 选择单药服用丙戊酸钠的癫痫患儿131例,应用多重PCR方法对CYP2A6*4基因多态性进行检测,应用PCR-连接酶检测反应技术对CYP2 B6*6、CYP2C9*2、CYP2C9*3、CYP2C19*2和CYP2C19*3基因多态性进行检测,应用均相酶放大免疫分析法测定丙戊酸钠血药浓度,采用单因素方差分析方法或t检验进行统计学分析.结果 患儿根据CYP2C9、CYP2C19基因型分为4组:G1组(CYP2C9和CYP2C19均为强代谢者)、G2组(CYP2C19中间代谢者)、G3组(CYP2C19弱代谢者)和G4(CYP2C9弱代谢者);G3(3.70±0.95)、G4组(4.35±1.48)标准化血药浓度显著高于G1组(2.57±1.30,t=3.056、4.490,均P<0.01)和G2组(2.76±1.19,t=2.827、4.462,均P<0.01);G3(19.46±5.20)、G4组(19.30 ±7.67)丙戊酸钠剂量(mg/d)显著低于G1组(24.10±6.97,t=2.359、2.297,均P<0.05).未发现突变型CYP2A6*4和CYP2B6*6对丙戊酸钠剂量和丙戊酸钠标准化血药浓度的影响.结论 CYP2C9和CYP2C19基因多态性会影响丙戊酸钠血药浓度,弱代谢(G3和G4组)的患儿服用丙戊酸钠应适当减少剂量.
[本文引用:1]
|
| [43] |
|
| [44] |
丙戊酸作为一种广谱抗癫痫药物在临床使用广泛。除用于癫痫治疗外,丙戊酸还常被用于双相障碍、精神分裂、人格障碍等疾病的治疗。目前有关丙戊酸遗传药理学的研究已引起众多国内外学者的关注,已有多项研究证实遗传因素在丙戊酸的代谢和不良反应的发生中起重要作用。本文对今年来有关丙戊酸遗传药理学方面的研究进展情况进行了整理、分析和讨论,为进一步开展丙戊酸的遗传药理学研究提供参考。
[本文引用:1]
|
| [45] |
Valproic acid (VPA) is a widely used anticonvulsant that is also approved for mood disorders, bipolar depression, and migraine. In vivo, valproate is metabolized oxidatively by cytochromes P450 and 尾-oxidation, as well as conjugatively via glucuronidation. The acyl glucuronide conjugate (valproate-glucuronide or VPAG) is the major urinary metabolite (30鈥50% of the dose). It has been hypothesized that glucuronidation of antiepileptic drugs is spared over age, despite a known decrease in liver mass. The formation rates of VPAG in a bank of elderly (65 years onward) human liver microsomes (HLMs) were measured by liquid chromatography/tandem mass spectrometry and compared with those in a younger (2鈥56 years) HLM bank. In vitro kinetic studies with recombinant UDP-glucuronosyltransferases (UGTs) were completed. A 5- to 8-fold variation for the formation of VPAG was observed within the microsomal bank obtained from elderly and younger donors. VPAG formation ranged from 6.0 to 53.4 nmol/min/mg protein at 1 mM substrate concentration (n = 36). The average velocities at 0.25, 0.5, and 1 mM VPA were 7.0, 13.4, and 25.4 nmol/min/mg protein, respectively, in the elderly HLM bank. Rates of VPAG formation were not significantly different in the HLM bank obtained from younger subjects. Intrinsic clearances (Vmax/Km) for several cloned, expressed UGTs were determined. UGT1A4, UGT1A8, and UGT1A10 also were found to catalyze the formation of VPAG in vitro. This is the first reported activity of these UGTs toward VPA glucuronidation. UGT2B7 had the highest intrinsic clearance, whereas UGT1A1 demonstrated no activity. In conclusion, our investigation revealed no differences in VPAG formation in younger versus elderly HMLs and revealed three other UGTs that form VPAG in vitro.
[本文引用:1]
|
| [46] |
Glucuronide conjugation of xenobiotics containing a carboxylic acid moiety represents an important metabolic pathway for these compounds in humans. Several human UDP-glucuronosyltransferases (UGTs) have been shown to catalyze the formation of acyl-glucuronides, including UGT2B7, UGT1A3, and UGT1A9. In this study, recombinant expressed UGT isoforms were investigated with many structurally related carboxylic acid analogues, and the UGT rank order for catalyzing the glucuronidation of carboxylic acids was UGT2B7026202UGT1A302≈02UGT1A9. Despite being a poor substrate with UGT1A3, coumarin-3-carboxylic acid was not a substrate for any other UGT isoform tested in this study, suggesting that it could be a specific substrate for UGT1A3. Interestingly, UGT1A7 and UGT1A10 also react with several carboxylic acid aglycones. Kinetic analysis showed that UGT2B7 exhibits much higher glucuronidation efficiency ( V max / K m ) with ibuprofen, ketoprofen, and others, compared to UGT1A3. These data indicate that UGT2B7 could be the major isoform involved in the glucuronidation of carboxylic acid compounds in humans.
[本文引用:0]
|
| [47] |
Valproic acid glucuronidation kinetics were carried out with three human UGT isoforms: UGT1A6, UGT1A9, and UGT2B7 as well as human liver and kidney microsomes. The glucuronidation of valproic acid was typified by high K m values with microsomes and expressed UGTs (2.3–5.202mM). The ability of valproic acid to interact with the glucuronidation of drugs, steroids and xenobiotics in vitro was investigated using the three UGT isoforms known to glucuronidate valproic acid. In addition to this the effect of valproic acid was investigated using two other UGT isoforms: UGT1A1 and UGT2B15 which do not glucuronidate valproic acid. Valproic acid inhibited UGT1A9 catalyzed propofol glucuronidation in an uncompetitive manner and UGT2B7 catalyzed AZT glucuronidation competitively ( K i =1.6±0.0602mM). Valproate significantly inhibited UGT2B15 catalyzed steroid and xenobiotic glucuronidation although valproate was not a substrate for this UGT isoform. No significant inhibition of UGT1A1 or UGT1A6 by valproic acid was observed. These data indicate that valproic acid inhibition of glucuronidation reactions is not always due to simple competitive inhibition of substrates.
[本文引用:0]
|
| [48] |
Valproic acid (VPA) is a widely used anticonvulsant that is also approved for mood disorders, bipolar depression, and migraine. In vivo, valproate is metabolized oxidatively by cytochromes P450 and 尾-oxidation, as well as conjugatively via glucuronidation. The acyl glucuronide conjugate (valproate-glucuronide or VPAG) is the major urinary metabolite (30鈥50% of the dose). It has been hypothesized that glucuronidation of antiepileptic drugs is spared over age, despite a known decrease in liver mass. The formation rates of VPAG in a bank of elderly (65 years onward) human liver microsomes (HLMs) were measured by liquid chromatography/tandem mass spectrometry and compared with those in a younger (2鈥56 years) HLM bank. In vitro kinetic studies with recombinant UDP-glucuronosyltransferases (UGTs) were completed. A 5- to 8-fold variation for the formation of VPAG was observed within the microsomal bank obtained from elderly and younger donors. VPAG formation ranged from 6.0 to 53.4 nmol/min/mg protein at 1 mM substrate concentration (n = 36). The average velocities at 0.25, 0.5, and 1 mM VPA were 7.0, 13.4, and 25.4 nmol/min/mg protein, respectively, in the elderly HLM bank. Rates of VPAG formation were not significantly different in the HLM bank obtained from younger subjects. Intrinsic clearances (Vmax/Km) for several cloned, expressed UGTs were determined. UGT1A4, UGT1A8, and UGT1A10 also were found to catalyze the formation of VPAG in vitro. This is the first reported activity of these UGTs toward VPA glucuronidation. UGT2B7 had the highest intrinsic clearance, whereas UGT1A1 demonstrated no activity. In conclusion, our investigation revealed no differences in VPAG formation in younger versus elderly HMLs and revealed three other UGTs that form VPAG in vitro.
[本文引用:1]
|
| [49] |
The aim of this study was to investigate the distribution and frequency of genetic polymorphisms in uridine diphosphate glucuronosyltransferase-2B7 (UGT2B7) in epilepsy patients and to evaluate the effect of these on the metabolism of valproic acid (VPA). Single nucleotide polymorphisms in UGT2B7 were investigated in 102 epilepsy patients using DNA sequencing and polymerase chain reaction–restriction fragment length polymorphism analysis. The steady-state plasma concentrations of VPA were determined in these patients, who had received VPA (approx. 500–1000mg/day) for at least 2 weeks. Fourteen patients had the CC genotype at UGT2B7 C802T, 46 carried CT, and 42 carried the TT genotype. At UGT2B7 G211T, 78 patients had the GG genotype, 23 carried GT, and one individual had the TT genotype. The standardized trough plasma concentration of VPA was much lower in those patients with a T allele at UGT2B7 C802T than in those with the CC genotype (TT, 2.11±1.26; CT, 2.31±1.25; CC, 3.02±1.32μgkgmL611mg611, p<0.01). However, UGT2B7 G211T polymorphisms had no influence on the plasma concentration of VPA (GG, 2.28±1.32, GT, 2.303±1.38μgkgmL611mg611). These results suggested that UGT2B7 C802T may be an important determinant of individual variability in the pharmacokinetics of VPA and that it may be necessary to increase the VPA dose for individuals with a T allele in order to achieve the therapeutic range of 50–100μg/mL.
[本文引用:1]
|
| [50] |
Glucuronidation by UDP-glucuronosyltransferase 2B7 (UGT2B7) has been identified as an important pathway for the elimination of its substrate drugs in humans. Alterations in UGT2B7 function or expression may influence individual variations in drug responses. In an effort to screen for UGT2B7 single nucleotide polymorphisms (SNPs) in Koreans, the UGT2B7 gene was directly sequenced in 50 normal subjects. A total of 19 genetic variations were found: seven in exons, eight in introns, and four in the 5'-untranslated region. The order of the frequency distribution of U GT2B7 variations was : – 900A >G, – 327G > A, 61161C>T, 10539A>G, 10711G>C and 10806T>A (40%); 2099T>A, 2100C>T, 2283A>G and 2316A>G (39%); 12029T>A (37%); 10928C>A (33%); 10541G>A (28%); 10897insA (24%); 372A > G (13%) and 211G > T (12%), as well as other minor alleles with less than 10% frequency. Nineteen variations were used to characterize linkage disequilibrium (LD) structures at the UGT2B7 locus. Eight tagging SNPs in UGT2B7 were determined. Identification of UGT2B7 SNPs with LD and the tagging SNPs lays the foundation for investigating UGT2B7-related genotype/phenotype association studies for Koreans as well as other populations.
[本文引用:1]
|
| [51] |
These results suggest that the haplotype structure in the Japanese population is different from that of other ethnic groups.
[本文引用:1]
|
| [52] |
In our previous study it was observed that the frequencies of UGT1A1*6, UGT2B7*3 and CYP2D6*10 in patients who have a low level ability of glucuronidation were significantly higher than those in patients with a high level of ability of glucuronidation. The same tendency was found in the frequency of CYP2D6*5, though there was no significant difference. The purpose of this study was to evaluate the effects of the polymorphism on pharmacokinetics of carvedilol by population pharmacokinetic analysis. Population pharmacokinetic analysis was performed using 373 plasma concentrations from 41 patients with chronic heart failure or angina pectoris. A one compartment pharmacokinetic model with first-order absorption (for oral dosing) was used to describe the concentration-versus-time data for carvedilol. We examined the effects of various clinical and genetic covariables in the regression models for clearance and volume of distribution. The results suggested that the factors of interindividual variation for carvedilol clearance were creatinine clearance and polymorphisms of UGT2B7 and CYP2D6 in the Japanese population with heart disease. It was estimated that UGT2B7*3 decreased the clearance of carvedilol by 37%, but UGT2B7*2 did not show any effect. Clearance in the patients who have intermediate activity of CYP2D6 was decreased by 39%.
[本文引用:1]
|
| [53] |
<p><strong>目的: </strong>探讨UGT2B7 A268G和UGT2B7 G211T基因多态性对丙戊酸血药浓度的影响。<strong>方法: </strong>用限制性片段长度多态性聚合酶链反应(polymerase chain reaction restriction fragment length polymorphism,PCR-RFLP)的方法分别对248名癫痫患者检测UGT2B7 A268G和UGT2B7 G211T两个位点的基因型,并收集患者的基本流行性病学资料和相关临床信息,如癫痫类型、病史、服药剂量、疗效和肝肾功能等。采用SPSS 13.0软件分别对资料进行多元线性回归、单因素方差分析、卡方检验和配对T检验等方法的数据统计。<strong>结果: </strong>多元线性回归分析显示,性别、年龄和体质量指数与丙戊酸血药浓度无明显相关,而浓度剂量比率则与血药浓度相关。在被纳入的248名患者中,UGT2B7 A268G和UGT2B7 G211T的基因型均符合哈迪温伯格平衡定律。UGT2B7-268A等位基因频率为30.05%,而G等位基因频率为69.95%,且携带AA,AG,GG不同基因型的患者服用丙戊酸后血药浓度的比较差异有统计学意义(<em>F</em>=5.477,<em>P</em>=0.005),AA基因组显著高于GG基因组(<em>P</em>=0.048),其他两组间比较差异无统计学意义(<em>P</em>>0.05).UGT2B7 211G等位基因频率为77.24%,T等位基因频率为22.58%;而携带该位点GG,GT和TT不同基因型的患者服用丙戊酸后,三者血药浓度间比较,差异无统计学意义(<em>P</em>>0.05)。<strong>结论: </strong>本研究揭示了UGT2B7A268G和UGT2B7G211T的基因多态在中国汉族癫痫人群中的分布,UGT2B7 A268G基因多态参与体内丙戊酸代谢并进而影响其血药浓度。临床上针对癫痫患者给予丙戊酸药物时,需要考虑患者携带UGT2B7A268G位所产生的影响而适当调整患者用药剂量。</p>
[本文引用:1]
|
| [54] |
Abstract AIMS: Valproic acid (VPA) is one of the most widely used antiepileptic drugs. The aim of the study was to investigate whether polymorphisms in genes related to pharmacokinetic and pharmacodynamic pathways of VPA were associated with the large interindividual variability in dosages and concentrations. METHODS & RESULTS: Genetic polymorphisms in six candidate genes were detected in 162 epileptic patients under maintenance with VPA monotherapy and stable seizure control by real-time PCR and PCR-RFLP. Results of statistical analysis demonstrated that carriers of the variant UGT1A6 19T>G, 541A>G and 552A>C allele tended to require higher VPA dosages and lower ln(concentration-to-dose ratios [CDRs]) than noncarriers (p G allele were more likely to require lower VPA dosages than noncarriers (p < 0.0001) and the homozygous carriers also tended to require lower dosages and higher lnCDRs (p < 0.0001). In addition, the regression model of CDR of VPA also revealed that genetic variants in UGT1A6, GRIN2B and UGT2B7 genes interactively affect CDRs of VPA (adjusted r脗虏 = 47%). CONCLUSION: Although there was a limited sample size, the study identified genetic factors associated with VPA therapy optimization that has not been revealed, and provided useful information for individualized VPA therapy in epileptic patients.
[本文引用:1]
|
| [55] |
Abstract Valproic acid (VPA) is one of the most commonly prescribed drugs for the treatment of epilepsy. Interindividual variability in VPA dose and plasma concentration may reflect functional consequences of genetic polymorphisms in genes encoding drug-metabolizing enzymes. The aim of this study was to determine the relationship between plasma concentrations of VPA and single nucleotide polymorphisms (SNPs) involving uridine diphosphate glucuronosyltransferase (UGT) 1A6 (UGT1A6), UGT2B7, and cytochrome P450 2C9 (CYP2C9) genes in Chinese children with epilepsy. UGT1A6, UGT2B7, and CYP2C9 polymorphisms were identified by the polymerase chain reaction-restriction fragment length polymorphism approach or direct automated DNA sequencing in 98 epileptic patients treated with VPA monotherapy. Patients with double heterozygosities at nucleotide positions T19G, A541G and A552C in the UGT1A6 gene, were associated with higher VPA doses compared to those with wild type or single heterozygosity (p = 0.010). Lower adjusted plasma VPA concentrations were also observed in patients with UGT1A6 double heterozygosities than those with single heterozygosity (p = 0.027). There were no differences in VPA dose or adjusted plasma VPA concentrations among the UGT2B7*2 or CYP2C9*3 genotypic groups. These results suggest that UGT1A6 mutations affect VPA metabolism in epileptic children. It needs to be further investigated in a larger cohort of patients.
[本文引用:2]
|
| [56] |
Purpose The aim of this study was to investigate the genetic polymorphisms of UGT1A3, UGT1A6, and UGT2B7 in Chinese epilepsy patients and their potential influence on the pharmacokinetics of valproic acid (VPA). Methods The genetic architectures of UGT1A3, UGT1A6, and UGT2B7 in 242 epilepsy patients were detected by DNA sequencing and PCR-restriction fragment length polymorphism. Steady-state plasma concentrations of VPA in 225 patients who had received VPA (approx. 250–1,00002mg/day) for at least 2 weeks were determined and associated with UGT polymorphisms. Results The allelic distribution of UGT1A3 in our Chinese epilepsy patients was significantly different from that in healthy subjects based on reference data. The standardized trough plasma concentration (C S ) of VPA was much lower in our patients with the UGT1A3*5 variant than in the wild type carriers (3.2465±651.05 vs. 4.6865±651.2402μg·kg·mL -1 ·mg -1 , P 65<650.01). UGT polymorphisms had no influence on the pharmacokinetic interactions between carbamazepine and VPA. Conclusion Our results suggest that UGT1A3*5 may be an important determinant of individual variability in the pharmacokinetics of VPA and that it may be necessary to increase VPA dose for UGT1A3*5 carriers to ensure its therapeutic range of 50–10002μg/mL.
[本文引用:3]
|
| [57] |
Two missense mutations were uncovered in the UGT1A6 (HLUG P1) cDNA which codes for a human phenol-metabolizing UDP-glucuronosyltransferase. The mutant and a wild-type UGT1A6 cDNAs were isolated from a custom synthesized human liver lambda Zap cDNA library. Both an A to G transition at nucleotide 541 (T181 A) and an A to C transversion at nucleotide 552 (R184S) occurred in exon 1 of the UGT1A6 (UGT1F) gene at the UGT1 locus. The two mutations on a single allele created a heterozygous genotype. Newly created BsmI and BsoFI sites at the T181 A and R184S locations, respectively, were confirmed by endonuclease treatment of PCR-generated DNA using the donor-liver genomic DNA as template. Screens with endonuclease treatment showed that 33/98 DNA samples were heterozygous with both mutations on one allele. One other individual also carried the R184S mutation on the second allele. Wild-type UGT1A6 generated a broad plateau of activity from pH 5.0 to pH 8.0 with certain experimental phenols, while activity was 1.3-2.5-fold higher at pH 6.4 than at pH 7.2 for others. UGT1A6*2 (181 A+ and 184S+) metabolized 4-nitrophenol, 4-tert-butylphenol, 3-ethylphenol/4-ethylphenol, 4-hydroxycoumarin, butylated hydroxy anisole and butylated hydroxy toluene, with the pH 6.4 preference, at only 27-75% of the rate of the wild-type isozyme whereas 1-naphthol, 3-iodophenol, 7-hydroxycoumarin, and 7-hydroxy-4-methylcoumarin were metabolized at essentially the normal level. Furthermore, UGT1A6*2 metabolized 3-O-methyl-dopa and methyl salicylate at 41-74% of that of the wild-type, and a series of beta-blockers at 28-69% of the normal level. This evidence suggests that the UGT1A6 enzyme activity is affected by different amino acids depending upon the substrate selection.
[本文引用:1]
|
| [58] |
目的:检测尿苷二磷酸葡糖醛酰转移酶(UGT)1A6 A541G 基因多态性在汉族癫癎患儿中的分布和突变频率,探讨UGT1A6 A541G突变各基因型与丙戊酸血药浓度的关系。方法:气相色谱法测定丙戊酸血药浓度,PCR-RFLP技术检测UGTIA6 A541G基因多态性,PCR扩增产物直接测序验证基因型检测方法的可靠性。结果:147例汉族癫癎患儿中UGTIA6 541位点的基因型AA、AG、GG分别为76例、65例和6例。服用单位剂量(mg/kg)引起的血药浓度AA基因型患儿为 3.91±1.57 μg/mL, AG基因型患儿为3.59±1.39 μg/mL, GG基因型患儿为3.73±1.28 μg/mL。AG、GG基因型患儿血药浓度较AA基因型患儿偏低,但差异无统计学意义。结论:UGT1A6 A541G基因多态性与丙戊酸的血药浓度无显著相关性,临床上个体血药浓度的差异可能是多种因素共同作用的结果。[中国当代儿科杂志,2010,12(6):429-432]
[本文引用:1]
|
| [59] |
|
| [60] |
Metabolism of valproic acid, a widely used drug, is only partially understood. It is mainly metabolized through glucuronidation and acts as a substrate for various UDP-glucuronosyltransferases (UGTs). UGTs metabolizing valproic acid in the liver are UGT1A3, UGT1A4, UGT1A6, UGT1A9 and UGT2B7, with UGT1A6 and UGT2B7 being the most prominent. Polymorphisms in genes expressing these enzymes may have clinical consequences, regarding dosing, blood levels of the drug and adverse reactions. Not all genes are well studied and studies, where they exist, report conflicting results. Prevalence of polymorphisms and various haplotypes is also of great importance, as it may suggest different therapeutic approaches in various populations. Presented here is a review of currently known polymorphisms, their functional impact, when known, and their prevalence in different populations, highlighting the current state of understanding and areas where there is a lack of data and suggesting new perspectives for further research.
[本文引用:1]
|
| [61] |
DOI:10.1345/aph.1D374
URL
[本文引用:1]
|
| [62] |
To present, compare, and contrast the various approaches to estimating population pharmacokinetic (PPK) models with respect to the mathematical foundation, statistical aspects, software programs for implementation, and underlying assumptions.Information on PPK was retrieved from a MEDLINE search (1977-August 2004) of literature and a bibliographic review of review articles and books. This information is used in conjunction with experience to explain the various methodologic approaches to PPK.All articles indentified from data sources were evaluated and relevant information was included in this review.Over 80 articles dealing with PPK estimation methods and/or their implementation were identified and reviewed. Sixty-four of these were chosen for their direct relevance to the subject of this article. Different estimation methods ranging from the naive averaging and naive pooled approaches through the standard two-stage approach to the nonlinear mixed-effects modeling approaches for estimating PPK are reviewed with their advantages and limitations.PPK estimation methods that rely on the characterizing of mixed (fixed and random) effects are known to produce PPK parameter estimates that are less biased than those obtained using the naive and standard two-stage approaches. The NONMEM software is the most widely used software for the characterization of PPK.
[本文引用:0]
|
| [63] |
OBJECTIVE: To present a framework within which population pharmacokinetic (PPK) studies should be designed and analyzed and discuss the application of developed PPK models. METHODS: Information on PPK was retrieved from a MEDLINE search (1979-December 2003) of the literature and a bibliographic evaluation of review articles and books. This information is used in conjunction with experience to explain the design and analysis of PPK studies. Also, examples are included to demonstrate the usefulness of PPK. SYNTHESIS: A great deal of thought must be given to the design and analysis of PPK studies (ie, development of PPK models). Models are of 2 primary types--descriptive and predictive--and the process applied to these models is necessarily different. An approach that ensures model applicability is presented. CONCLUSIONS: PPK models have great utility, and the applications are many. They are very different from single-subject pharmacokinetic models and therefore require different approaches to model estimation.
[本文引用:1]
|
| [64] |
Population pharmacokinetics (PPK) has evolved from a discipline primarily applied to therapeutic drug monitoring to one that plays a significant role in clinical pharmacology in general and drug development in particular. In February 1999 the US Food and Drug Administration issued a 鈥楪uidance for Industry: Population Pharmacokinetics鈥 that sets out the mechanisms and philosophy of PPK and outlines its role in drug development.The application of PPK to the drug development process plays an important role in the efficient development of safe and effective drugs. PPK knowledge is essential for mapping the response surface, explaining subgroup differences, developing and evaluating competing dose administration strategies, and as an aid in designing future studies. The mapping of the response surface is done to maximise the benefit-risk ratio, so that the impact of the input profile and dose magnitude on beneficial and harmful pharmacological effects can be understood and applied to individual patients. PPK combined with simulation methods provides a tool for estimating the expected range of concentrations from competing dose administration strategies. Once extracted, this knowledge can be applied to labelling or used to assess various future study designs.PPK should be implemented across all phases of drug development. For preclinical studies, PPK can be applied to allometric scaling and toxicokinetic analyses, and is useful for determining 鈥榝irst time in man鈥 doses and explaining toxicological results. Phase I studies provide initial understanding of the structural model and the effect of possible covariates, and may later be used to evaluate PPK differences between patients and healthy individuals. Phase II studies provide the greatest opportunity to map the response surface. With these PPK models it is possible to gain an improved understanding of the role of the dose on the response surface and of the range of expected responses. In phase III and IV studies, PPK is implemented to further refine the PPK model and to explain unexpected responses.Planning for the implementation of PPK across all phases of drug development is necessary, as well as planning for individual PPK studies. Planning should include: defining important questions, identifying covariates and drug-drug interactions that need to be investigated, and identifying the applications and intended use of the model(s). The plan for each project must have a strategy for data management, data collection, data quality assurance, staff training for data collection, data analysis and model validation.
[本文引用:1]
|
| [65] |
|
| [66] |
UDP-glucuronosyltransferases (UGTs) are critical for the metabolism and clearance of drugs, chemicals, and hormones. The development of UGT1A1 and 1A6 was studied in 50 pediatric liver samples using bilirubin, serotonin activity assays, and Western blot as well as pharmacokinetic scaling. UGT activity developed age dependently in pediatric liver. Maximal activity of 0.7690 nmol · min · (-1) mg protein(-1) was observed for UGT1A1 at 3.8 months. For UGT1A6, activity matured at 14 months (4.737 nmol · min · (-1)mg protein(-1)). Protein expression was not age-dependent, and activities did not correlate to protein levels for either enzyme. The in vitro activities were used to calculate normalized hepatic clearances using both allometric scaling and a physiologically based pharmacokinetic model. For UGT1A1, allometry predicted normalized adult clearances of 0.0070 l · h(-1) · kg(-1) at 3.0 (well stirred) and 2.8 years (parallel tube), whereas the Simcyp model showed normalized clearances of 0.0079 l · h(-1) · kg(-1) at 2.6 (well stirred) and 2.5 years (parallel tube). For UGT1A6, only the Simcyp well stirred model converged at 0.3524 l · h(-1) · kg(-1) at 12.6 months. These data imply independent regulation of UGT1A1 and 1A6 where activity has matured after 6 months to 1 year. Total hepatic clearance of substances mediated by these enzymes may mature concurrently or take longer because of other physiological factors. Late development of UGT enzymes may contribute to chemical, drug, and environmental toxicity.
[本文引用:1]
|
| [67] |
|
| [68] |
Valproic acid glucuronidation kinetics were carried out with three human UGT isoforms: UGT1A6, UGT1A9, and UGT2B7 as well as human liver and kidney microsomes. The glucuronidation of valproic acid was typified by high K m values with microsomes and expressed UGTs (2.3–5.202mM). The ability of valproic acid to interact with the glucuronidation of drugs, steroids and xenobiotics in vitro was investigated using the three UGT isoforms known to glucuronidate valproic acid. In addition to this the effect of valproic acid was investigated using two other UGT isoforms: UGT1A1 and UGT2B15 which do not glucuronidate valproic acid. Valproic acid inhibited UGT1A9 catalyzed propofol glucuronidation in an uncompetitive manner and UGT2B7 catalyzed AZT glucuronidation competitively ( K i =1.6±0.0602mM). Valproate significantly inhibited UGT2B15 catalyzed steroid and xenobiotic glucuronidation although valproate was not a substrate for this UGT isoform. No significant inhibition of UGT1A1 or UGT1A6 by valproic acid was observed. These data indicate that valproic acid inhibition of glucuronidation reactions is not always due to simple competitive inhibition of substrates.
[本文引用:1]
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
A population analysis of the kinetics of valproic acid (VPA) in children with epilepsy was performed in order to characterize the covariates which influence VPA clearance (CL).A total of 770 steady-state serum concentration samples was analysed. These were collected during VPA therapy from 255 children, aged 0.1-14 years and weighting 4-74 kg. Age, total body weight (TBW), VPA daily dose, sex and comedication with carbamazepine (CBZ) were considered as covariates. Population analysis was made with NONMEM program, assuming a one-compartment model, fixing the VPA absorption rate, bioavailability and distribution volume at values found in the literature. The results of the population pharmacokinetics analysis were validated in a group of 45 epileptic patients.The final regression model for VPA clearance, that included TBW (kg), daily dose (mg/kg) and CBZ comedication as covariates with a significant influence on this parameter, was as follows: CL (L/h) = 0.012 TBW0.715 DOSE0.306(1.359 CBZ). The coefficient of variation for interpatient variability in CL was 21.4% and the residual variability estimated was 23.9% for a concentration of 65 mg/l. In order to estimate the predictive performance of the selected final model, predictions of the VPA serum concentrations were calculated and compared with VPA measured concentrations in the validation group. This assessment revealed an important improvement in the predictive performance of VPA concentrations in comparison with the basic model that did not include any covariates (root squared mean error: 19.50 vs. 39.73 mg/l).A population pharmacokinetic model is proposed to estimate the individual CL for paediatric patients receiving VPA in terms of patient's dose, weight and concomitant CBZ, in order to establish a priori dosage regimens.
[本文引用:1]
|
| [73] |
BACKGROUND: The aim of this study was to determine the factors that influence valproate clearance (CL) in Mexican epileptic pediatric patients using a mixed-effect model and sparse data of serum concentrations of valproic acid (VPA) collected during routine clinical care of patients. METHODS: The number of patients included in the study was 110. The population CL was calculated by using the NONMEM program. The following covariates were tested by their influence on CL: total body weight (TBW), height, age, body surface area, daily dose (DD), sex of the patient and comedication with phenobarbital (PB) or carbamazepine. RESULTS: The final regression model for valproic CL found best to describe the data was: CL/F=(0.0466+0.00363 TBW+0.000282 DD)*(1+0.236 PB). This model allows a reduction of 50% of the interindividual variability and of 31% of the residual variability described by the basic model that does not include covariables. CONCLUSIONS: Total body weight, daily dose of valproate and concomitant therapy with PB are factors that significantly influence VPA kinetic disposition and they should be considered in programming dosage regimens for this antiepileptic drug in the pediatric population. The validation of the model supports its acceptability for clinical purposes.
[本文引用:1]
|
| [74] |
Abstract The aim of the present study was to estimate valproic acid (VPA) clearance values for adult patients with epilepsy, using serum concentrations gathered during their routine clinical care. Retrospective steady state serum concentrations data ( n =534) collected from 208 adult patients receiving VPA were studied. Data were analysed according to a one-compartment model using the NONMEM program. The influence of VPA daily dose (Dose), gender, age, total body weight (TBW), and comedication with carbamazepine (CBZ), phenytoin (PHT) and phenobarbital (PB) were investigated. The results of the population pharmacokinetics analysis were validated in a group of 30 epileptic patients. The final regression model for VPA clearance (Cl) was: (1) The inter-individual variability in VPA clearance, described by a proportional error model, had a variation coefficient (CV) of 23.4% and the residual variability, described using an additive model, was 11.4 mg/L. These results show that VPA clearance increased linearly with TBW, but increases nonlinearly with increasing VPA daily dose. Concomitant administration of CBZ, PHT and PB led to a significant increase in VPA clearance. The model predictions in the validation group were found to have satisfactory precision and bias. In conclusion, inter-individual variability in VPA clearance can be partly explained by TBW, daily dose and bitherapy with CBZ, DPH or PB. Inclusion of these factors allows this variability to be reduced by 37.23% which may be very useful for clinicians when establishing the initial VPA dosage regimen. However, the magnitude of inter-individual plus residual variabilities, remaining in the final model, render these dosage predictions imprecise and justify the need for VPA serum level monitoring in order to individualize dosage regimens more accurately. Copyright 漏 1999 John Wiley & Sons, Ltd.
[本文引用:1]
|
| [75] |
Abstract BACKGROUND:: There are several reports describing population pharmacokinetic (PPK) models of valproic acid (VPA).However, little was known in Chinese adult patients with epilepsy. The present study aimed to establish a PPK model for VPA in Chinese adult epilepsy patients and to demonstrate its use for dose individualisation. METHODS:: Data were obtained from a prospective study of 199 adult epilepsy patients at five hospitals. The trough concentrations at steady state were measured by fluorescence polarisation immunoassay (FPIA). Data were analysed using the Nonlinear Mixed Effects Model (NONMEM) software. The serum trough concentrations at steady state were also measured using samples (n=20) collected prospectively from a different hospital from those providing the data for deriving the original model. These independent samples served as an evaluation group. RESULTS:: The important determinants of apparent VPA clearance (CL/F) were daily dose, body weight, and combination with carbamazepine, phenytoin, or phenobarbital. The final model predicted the individualised doses accurately. A total of 85% of the trough concentrations in the evaluation group were accurately predicted by the final model, while the prediction errors of the other patients were all less than 卤31%. CONCLUSIONS:: A PPK model was developed to estimate the individual CL for patients taking VPA and could be applied for individualising doses in the target population.
[本文引用:1]
|
| [76] |
|
| [77] |
Abstract The objective of this study was to identify factors that affect valproic acid (VPA) apparent clearance (CL/F) in elderly nursing home residents. Inclusion criteria included residency in a nursing home for at least 2 months, aged 65 years or older, a stable dosing regimen of VPA for at least 4 weeks, VPA concentration, and complete dosing information. CL/F was analyzed by a nonlinear mixed effects model. A one-compartment model with first-order absorption and elimination was used. Both volume and absorption rate constant were fixed (14 L and 1 hr, respectively). Covariates were tested by forward inclusion and backward elimination. Interindividual variability in clearance was estimated using an exponential error model and expressed as a coefficient of variation. Residual error was estimated using a combined additive and constant coefficient of variation error model. The study consisted of 405 observations from 146 (52 men, 94 women) elderly nursing home residents. CL/F was not affected by age or weight. The population CL/F was 0.843 L/hr. CL/F was 1) 27% lower in female residents; 2) 41% greater when the resident was on concomitant metabolic inducers carbamazepine or phenytoin cotherapy; and 3) 25% greater when the syrup formulation was used. Variability in CL/F was 32.9%. Coefficient of variation and standard deviation of the residual error were 18.2% and 10.6 mg/L, respectively. The increased CL/F in patients taking VPA syrup may be the result of a decreased bioavailability (F) rather than an increased CL that could be associated with pathology requiring use of the syrup rather than an inherent property of the drug formulation. The results from this study may be useful for individualizing dose regimens in the nursing home population based on patient-specific factors.
[本文引用:1]
|
| [78] |
In the absence of enzyme inducing comedication, VPA clearance in the elderly was comparable to that observed in controls. VPA clearance in elderly patients receiving enzyme inducing AEDs was lower than in controls, the difference being probably due to an influence of age as well as to the fact that mean VPA dosage was lower in these patients than in controls. Since our measurements of clearance were based on total serum VPA concentrations and VPA binding to plasma proteins is known to be reduced in old age, it is likely that the clearance of unbound, pharmacologically active, VPA was decreased to an important extent in the elderly, presumably as a result of a decline in drug metabolizing capacity.
[本文引用:1]
|
| [79] |
Purpose To evaluate the effects of CYP2C19 and CYP2C9 genotypes on the pharmacokinetic variability of valproic acid (VPA) in epileptic patients using a population pharmacokinetic (PPK) approach. Methods VPA concentrations were measured in 287 epileptic patients, who were genotyped for CYP2C19*2/*3 and CYP2C9*3 . Patients who were on monotherapy with VPA were divided into two groups, a PPK-model group ( n 65=65177) and a PPK-valid group ( n 65=65110). The PPK parameter values for VPA were calculated in the PPK-model group by using the NONMEM software. Ultimately, a biological model and a final model were established. Each model was then used to independently predict the concentrations of the PPK-valid group to validate the two models. Results There was a significant effect of the CYP2C19 and CYP2C9 genotypes on the pharmacokinetic (PK) variability ( P65 <650.01) in the final PPK model of CL/F. The interindividual CL was calculated according to the final model: CL/F65=650.095165×65(165+65e 0.026765×65(3656165genotype) )65+650.007165×65age (L/h). The coefficient of variation (CV) (omega CL/F) of the final model was 29.3%, while that of the biological model was 31.7%. Based on the genotype, the individual PK parameters can be calculated more accurately than before. Conclusion The CYP2C19 and CYP2C9 genotypes significantly influenced the PK variability of VPA, as quantified by NONMEM software.
[本文引用:1]
|
| [80] |
目的建立丙戊酸(VPA)在癫痫患者中的群体药代动力学(PPK)模型,考察固定效应因素对VPA清除率(CL/F)的影响。方法回顾性收集贵州省人民医院111名癫痫患者VPA稳态血药浓度数据及相应的人口学、合并用药及CYP2A6基因型等资料,随机将患者分成建模组(74名)及验证组(37名),使用建模组数据通过非线性混合效应模型(NONMEM)程序建立VPA的PPK模型。使用验证组数据来验证模型的准确度和精密度,比较基础模型和最终模型的平均预测误差(MPE)、平均绝对误差(MAE)、平均根方差(RMSE)。结果建立的最终模型包含了日用药剂量(DDO)及CYP2A6基因型,模型方程为:CL/F=0.363.DDO0.525.1.29GENECYP2A6。最终模型有更好的精密度及准确度,基础模型MPE、MAE、RMSE值为-10.631、4.40、22.55,最终模型相应值为-6.11、9.06、14.17。结论本研究初步建立癫痫患者VPA的PPK模型,VPA清除率随日给药剂量的增大而增大,CYP2A6野生型(CYP2A6*1/*1)组患者较CYP2A6突变型(CYP2A6*1/*4、CYP2A6*4/*4)组患者有更高的VPA清除率。
[本文引用:1]
|
| [81] |
|

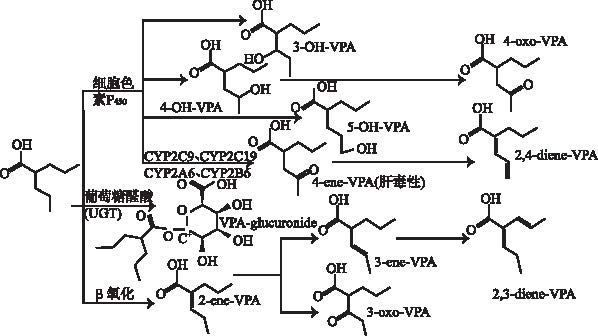
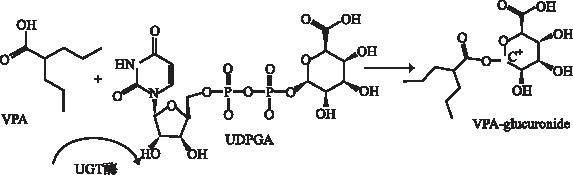
{kind=link}
{kind=link}
{kind=link}
{kind=link}