中国科技论文统计源期刊 中文核心期刊
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖

 , 常山泉
, CHANG Shanquan
, 常山泉
, CHANG Shanquan
抑郁症的发病机制非常复杂,至今尚未完全阐明。大量临床及临床前研究表明,5-羟色胺(5-HT)能神经功能障碍可能是导致抑郁症的关键因素之一。在5-HT神经系统中,除 5-羟色胺转运体(SERT)外,有多种5-HT受体亚型与抑郁症有关,尤以5-HT1A及5-HT2A受体与抑郁症关系最为密切。5-HT2A受体在大脑中广泛分布,是调节情绪的重要物质基础,对情绪、感知的调控具有重要作用。5-HT2A受体可以直接或间接调节单胺类递质释放,调节脑中单胺类递质水平,参与抑郁症发病过程。5-HT2A受体拮抗剂可以增强5-羟色胺再摄取抑制剂等抗抑郁药对难治性抑郁症的治疗效果并减少性功能障碍及睡眠障碍等不良反应。目前有不少以5-HT2A受体为靶点的抗抑郁药应用于临床,也有大量化合物处于临床及临床前研究。该文对5-HT2A受体与抑郁症的关系及以5-HT2A受体为靶点的抗抑郁药研究进展进行简要综述,以期为新型抗抑郁药物的研发提供参考。
The pathogenesis of depression is highly complicated and has not been fully elucidated.Numerous clinical and preclinical studies suggested that serotonergic (5-HT) neurological dysfunction critically contributed to depression.In addition to the serotonin transporter (SERT),multiple subtype receptors in the 5-HT nervous system are related to depression.Among them,5-HT1A and 5-HT2A receptors are most closely associated.Brain widely distributed 5-HT2A receptors provide a fundamental basis for regulating mood and perception.5-HT2A receptors modulate the level of monoamine transmitters in the brain by directly or indirectly regulating monoamine transmitters' release to participate in depression development.5-HT2A receptors antagonists enhanced the therapeutic effects of antidepressants such as selective serotonin reuptake inhibitors (SSRIs) for treatment-resistant depression and reduced adverse reactions such as sexual dysfunction and sleep disorder.Several 5-HT2A receptor-targeted antidepressants were approved for clinical treatment,and many compounds are currently being researched in clinical or preclinical studies.This review briefly discussed the relationship between 5-HT2A receptors and depression and summarized the research progress of antidepressants targeting 5-HT2A receptors to provide information for antidepressant research.
开放科学(资源服务)标识码(OSID)
抑郁症是一种以持续情绪低落、兴趣缺失、睡眠障碍、食欲不振及性欲低下为主要临床特征的精神障碍性疾病[1]。抑郁症发病机制非常复杂,至今尚未完全阐明。大量临床及临床前研究表明,5-羟色胺(5-hydroxytryptamine or serotonin,5-HT)能神经功能障碍可能是导致抑郁症的关键因素之一,去甲肾上腺素(noradrenaline or norepinephrine,NA or NE)能神经以及多巴胺(dopamine,DA)能神经也参与了抑郁症的发病过程[2]。目前临床使用的大部分抗抑郁药物,如选择性5-羟色胺再摄取抑制剂(selective serotonin reuptake inhibitor,SSRIs)、5-羟色胺及去甲肾上腺素双重再摄取抑制剂(serotonin-norepinephrine reuptake inhibitors,SNRIs)等均能发挥抗抑郁作用[3]。5-HT受体亚型比较复杂,根据结构、生物化学及药理学差异,5-HT受体分为7个家族(5-HT1-5-HT7),共14个亚型,除5-HT3受体为配体门控型离子通道外,其余都属于G蛋白偶联受体[4]。研究表明,5-HT受体介导包括焦虑症、强迫症和抑郁症等调节情感在内的多种生理功能[5,6]。其中,5-HT1A、5-HT1B、5-HT2A、5-HT2C、5-HT3、5-HT4、5-HT6及5-HT7等受体与抑郁症有关,尤以5-HT1A及5-HT2A受体与抑郁症关系最为密切[7]。有关 5-HT1A受体与抑郁症的关系及相关药物的研究进展已有较多的综述报道[8],但与5-HT2A受体相关的报道笔者较少见到。因此,笔者在本文对5-H
有文献报道,在抑郁症患者及抑郁症自杀患者尸检脑组织中皮质5-HT2A受体密度显著增加,这可能与抑郁症患者脑中5-HT水平较低、代偿性导致5-HT2A受体密度增加有关[9]。多种抗抑郁药物在长期服用后均可下调5-HT2A受体密度并通过影响前脑皮质下环路发挥抗抑郁作用,提示5-HT2A受体与抑郁症的发病及抗抑郁药关系密切[11]。鉴于新皮质中5-HT1A和5-HT2A受体的大量共表达,阻断5-HT2A受体可以增强5-HT1A受体介导的皮质和边缘区域神经传递,这种效应可能与抗抑郁相关[10]。此外,抑郁症发病的单胺假说认为脑中单胺类递质水平与抑郁症发病密切相关,而5-HT2A受体可以直接或间接调节单胺类递质释放,调节脑中单胺类递质水平,参与抑郁症发病及抗抑郁药物治疗[12]。
临床多数抑郁症患者对药物治疗或心理治疗有效,但仍有部分患者对各种药物均无反应,即通常所称的难治性抑郁症[13]。临床试验表明,非典型抗精神病药物能增强SSRIs对难治性抑郁症的疗效,其机制可能与其拮抗5-HT2A受体、逆转SSRIs对蓝斑神经元的抑制作用以及增加5-HT及NE的释放有关,提示拮抗5-HT2A受体可以增强SSRIs抗抑郁效果,提高对难治性抑郁症的疗效[14]。此外,有研究表明,对5-羟色胺转运体(serotonin transporter,SERT)及5-HT2A受体具有两重抑制作用的抗抑郁药萘法唑酮比传统只抑制SERT的SSRIs出现性功能障碍及睡眠障碍不良反应明显减少,说明拮抗5-HT2A受体可以减少SSRIs不良反应[15]。这些证据均表明拮抗5-HT2A受体可以增强抗抑郁药疗效并减少不良反应。因此,开发具有5-HT2A受体拮抗作用的多靶点抗抑郁药可能是治疗难治性抑郁症的方向之一。
目前有多个上市的抗抑郁药或抗精神分裂症药都能作用于5-HT2A受体,通过拮抗5-HT2A受体发挥抗抑郁作用。这些药物在抗抑郁的同时,往往还有抗焦虑、改善睡眠以及减少性功能障碍的作用,相对于其他无5-HT2A受体活性的药物表现出独特的治疗优势,特别适合抑郁伴焦虑及失眠的患者。这些药物相关信息见
表1 具有抗抑郁作用的5-HT2A受体相关药物
Tab.1 5-HT2A receptor-related drugs with antidepressant effects
米氮平药理作用机制较独特,主要能阻断突触前α2自身受体或异质受体,同时能阻断5-HT2A受体、5-HT2C受体和5-HT3受体[19]。阻断α2自身受体或异源性受体,可以增加NE和5-HT释放及其神经传递,改善抑郁症患者的情感症状和躯体症状。5-HT2受体兴奋可引发失眠、焦虑激越和性功能障碍,5-HT3受体兴奋则可引起恶心,米氮平可阻断这两个受体,减少与5-HT相关的不良反应,如焦虑、失眠、呕吐、恶心及性功能障碍等[20]。米氮平适用于各种抑郁症的急性期及维持期治疗,特别是治疗伴有睡眠障碍或焦虑障碍的抑郁症、伴有焦虑激越或焦虑躯体化的抑郁症患者。此外,大量临床研究表明,米氮平单用或联合其他抗抑郁药可有效治疗难治性抑郁症[21]。
除以上已上市的具有抗抑郁作用的5-HT2A受体相关药物,还有较多以5-HT2A受体为靶点的化合物处于临床研究或临床前研究,有望开发成抗抑郁新药。
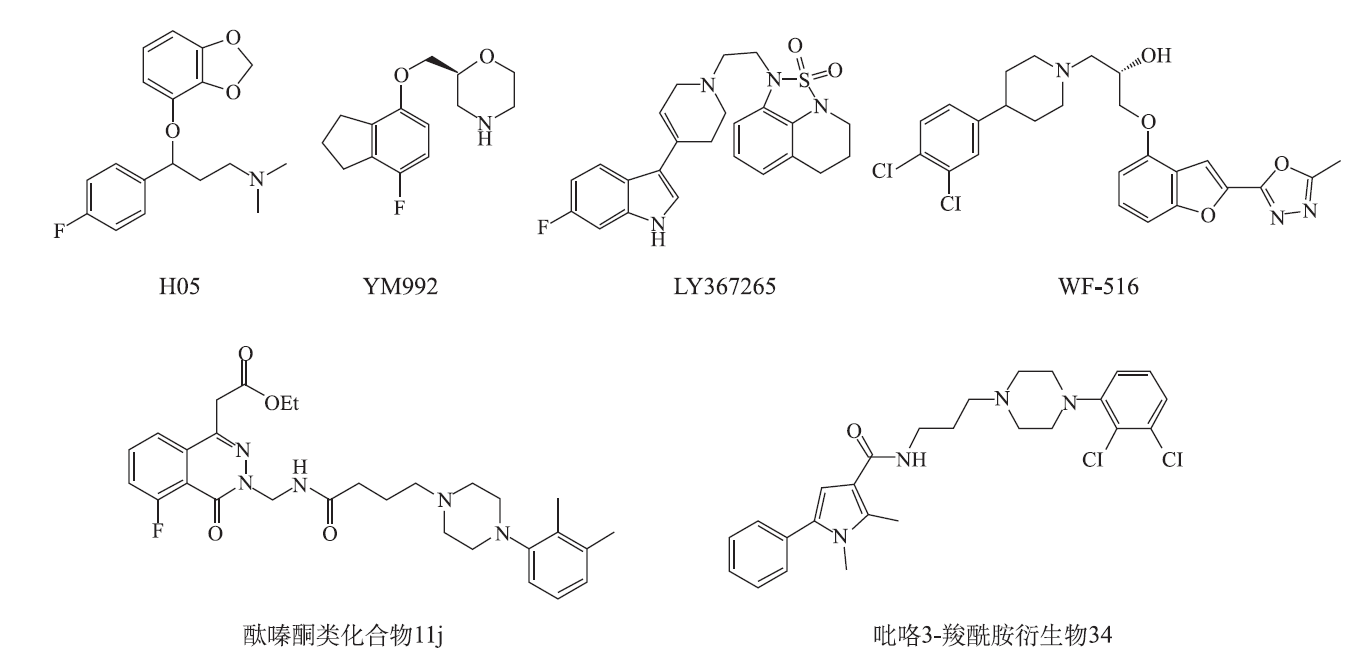
H05是一种新型3-(benzo[d][1,3]dioxol-4-yloxy)-3-arylpropyl amine衍生物(
YM992是HATANAKA等[33]筛选得到的一个对SERT(Ki=21 nmol·L-1)及5⁃HT2A受体(Ki=86 nmol·L-1)均有很强亲和力的化合物(图1)。体内试验表明,该化合物能明显增强5⁃羟色胺酸诱导的小鼠震颤、甩头等行为并减少2,5⁃二甲氧基⁃4⁃碘苯基丙烷⁃2⁃胺盐酸盐(4⁃iodo⁃2,5⁃dimethoxyphenyl⁃isopropylamine,DOI)诱导的小鼠甩头次数;小鼠悬尾试验结果表明,YM992及阿米替林均能明显减少小鼠悬尾不动时间。而西酞普兰及氟西汀虽有减少不动时间趋势,但差异无统计学意义,说明除5⁃HT再摄取抑制作用外,5⁃HT2A受体拮抗活性可能是YM992在小鼠悬尾试验中较西酞普兰及氟西汀显示出更强药效的原因。在大鼠嗅球摘除实验中,YM992单次及连续14 d给药均能明显改善模型大鼠被动回避学习障碍,但西酞普兰及阿米替林单次给药无效,只有14 d给药有效,说明SERT及5⁃HT2A受体双重抑制作用可能是YM992急性给药有效的机制之一[34]。
WF-516是一种研究中的抗抑郁化合物(
KIM等[38]合成的酞嗪酮类化合物11j(
此外,笔者通过检索Cortellis数据库发现,住友制药的DSP-1200、Intra-Cellular Therapies公司的ITI-1284、Biomind Labs公司的BMND-02以及江苏恩华药业的NH-102等新药都对5-HT2A受体具有较强拮抗活性,目前正处于临床研究不同阶段,拟用于抑郁症的治疗。
抑郁症是一类复杂的精神性疾病,其机制尚未完全阐明。目前抗抑郁药大多作用于单胺能神经系统,存在起效缓慢、临床效率不高及不良反应较多等诸多缺点[40]。从目前的研究结果来看,单一5-HT2A受体拮抗作用并不能产生明显的抗抑郁效果,需与其他抗抑郁靶点(如SERT、NET)协同作用才能达到增加药效或减少不良反应的效果。因此,目前开发以5-HT2A受体为靶点的抗抑郁药主要有两个策略:①开发多靶点的抗抑郁药,即一个药物能同时作用于包括5-HT2A受体在内的多个靶标,如米氮平、曲唑酮等;②开发复方药,如Symbyax,该药是礼来公司开发的奥氮平/盐酸氟西汀复方制剂,被FDA批准用于治疗双相抑郁症及难治性抑郁症。
此外,随着近年来对5-H
由于大脑是一个复杂的器官,其功能通常同时受很多神经或递质的调节,这些调节需达到一个精密的平衡,以维持脑的正常生理功能。因此,不管以哪种策略开发抗抑郁新药,都需要探索研究哪些靶标与5-HT2A受体能产生最佳的协同效应,以及与各靶标之间的最佳亲和力比值,以最大程度发挥5-HT2A受体增加抗抑郁药效及减少不良反应的作用。
| [1] |
Depressive disorder is one of the most widespread forms of psychiatric pathology, worldwide. According to a report by the World Health Organization, the number of people with depression, globally, is increasing dramatically with each year. Previous studies have demonstrated that various factors, including genetics and environmental stress, contribute to the risk of depression. As such, it is crucial to develop a detailed understanding of the pathogenesis of depressive disorder and animal studies are essential for identifying the mechanisms and genetic disorders underlying depression. Recently, many researchers have reported on the pathology of depression via various models of depressive disorder. Given that different animal models of depression show differences in terms of patterns of depressive behavior and pathology, the comparison between depressive animal models is necessary for progress in the field of the depression study. However, the various animal models of depression have not been fully compared or evaluated until now. In this paper, we reviewed the pathophysiology of the depressive disorder and its current animal models with the analysis of their transcriptomic profiles. We provide insights for selecting different animal models for the study of depression. © 2021 The Authors. CNS Neuroscience & Therapeutics Published by John Wiley & Sons Ltd.
DOI:10.1111/cns.13622
PMID:33650178
[本文引用:1]
|
| [2] |
|
| [3] |
|
| [4] |
Serotonin is a key neurotransmitter that is implicated in a wide variety of behavioral and cognitive phenotypes. Originating in the raphe nuclei, 5-HT neurons project widely to innervate many brain regions implicated in the functions. During the development of the brain, as serotonin axons project and innervate brain regions, there is evidence that 5-HT plays key roles in wiring the developing brain, both by modulating 5-HT innervation and by influencing synaptic organization within corticolimbic structures. These actions are mediated by 14 different 5-HT receptors, with region- and cell-specific patterns of expression. More recently, the role of the 5-HT system in synaptic re-organization during adulthood has been suggested. The 5-HT neurons have the unusual capacity to regrow and reinnervate brain regions following insults such as brain injury, chronic stress, or altered development that result in disconnection of the 5-HT system and often cause depression, anxiety, and cognitive impairment. Chronic treatment with antidepressants that amplify 5-HT action, such as selective serotonin reuptake inhibitors (SSRIs), appears to accelerate the rewiring of the 5-HT system by mechanisms that may be critical to the behavioral and cognitive improvements induced in these models. In this review, we survey the possible 5-HT receptor mechanisms that could mediate 5-HT rewiring and assess the evidence that 5-HT-mediated brain rewiring is impacting recovery from mental illness. By amplifying 5-HT-induced rewiring processes using SSRIs and selective 5-HT agonists, more rapid and effective treatments for injury-induced mental illness or cognitive impairment may be achieved.
[本文引用:1]
|
| [5] |
Depression is a polygenic and highly complex psychiatric disorder that remains a major burden on society. Antidepressants, such as selective serotonin reuptake inhibitors (SSRIs), are some of the most commonly prescribed drugs worldwide. In this review, we will discuss the evidence that links serotonin and serotonin receptors to the etiology of depression and the mechanisms underlying response to antidepressant treatment. We will then revisit the role of serotonin in three distinct hypotheses that have been proposed over the last several decades to explain the pathophysiology of depression: the monoamine, neurotrophic, and neurogenic hypotheses. Finally, we will discuss how recent studies into serotonin receptors have implicated specific neural circuitry in mediating the antidepressant response, with a focus being placed on the hippocampus.
DOI:10.1186/s13041-017-0306-y
PMID:28646910
[本文引用:1]
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
Serotonin modulates several physiological and cognitive pathways throughout the human body that affect emotions, memory, sleep, and thermal regulation. The complex nature of the serotonergic system and interactions with other neurochemical systems indicate that the development of depression may be mediated by various pathomechanisms, the common denominator of which is undoubtedly the disturbed transmission in central 5-HT synapses. Therefore, the deliberate pharmacological modulation of serotonergic transmission in the brain seems to be one of the most appropriate strategies for the search for new antidepressants. As discussed in this review, the serotonergic system offers great potential for the development of new antidepressant therapies based on the combination of SERT inhibition with different pharmacological activity towards the 5-HT system. The aim of this article is to summarize the search for new antidepressants in recent years, focusing primarily on the possibility of benefiting from interactions with various 5-HT receptors in the pharmacotherapy of depression.
[本文引用:2]
|
| [11] |
|
| [12] |
Serotoninergic neurons in the central nervous system impinge on many other neurons and modulate their neurotransmitter release. This review focuses on 1) the function of presynaptic 5-hydroxytryptamine (5-HT) heteroreceptors on axon terminals of central cholinergic, dopaminergic, noradrenergic, or GABAergic neurons and 2) the role of GABAergic interneurons expressing 5-HT heteroreceptors in the regulation of acetylcholine, dopamine, or noradrenaline release. In vitro studies on slices or synaptosomes and in vivo microdialysis experiments have shown that 5-HT(1A), 5-HT(1B), 5-HT(2A), 5-HT(2C), 5-HT(3), and/or 5-HT(4) heteroreceptors mediate this modulation. 5-HT(1B) receptors on neocortical cholinergic, striatal dopaminergic, or hippocampal GABAergic axon terminals are examples for release-inhibiting 5-HT heteroreceptors; 5-HT(3) receptors on hippocampal GABAergic or 5-HT(4) receptors on hippocampal cholinergic axon terminals are examples for release-facilitating 5-HT heteroreceptors. GABA released from GABAergic interneurons upon activation of facilitatory 5-HT receptors, e.g., 5-HT(2A) or 5-HT(3) receptors, mediates inhibition of the release of other neurotransmitters such as prefrontal neocortical dopamine or neocortical acetylcholine release, respectively. Conversely, attenuated GABA release in response to activation of inhibitory 5-HT heteroreceptors, e.g., 5-HT(1A) or 5-HT(1B) receptors on GABAergic interneurons is involved in paradoxical facilitation of hippocampal acetylcholine and striatal dopamine release, respectively. Such 5-HT heteroreceptors are considered potential targets for appropriate 5-HT receptor ligands which, by enhancing the release of a relevant neurotransmitter, can compensate for its hypothesized deficiency in distinct brain areas. Examples for such deficiencies are the impaired release of hippocampal or neocortical acetylcholine, striatal dopamine, and hippocampal or neocortical noradrenaline in disorders such as Alzheimer's disease, Parkinson's disease, and major depression, respectively.
PMID:18160701
[本文引用:1]
|
| [13] |
|
| [14] |
Treatment resistant depression (TRD) is associated with poor outcomes, but a consensus is lacking in the literature regarding which compound represents the best pharmacological augmentation strategy to antidepressants (AD). In the present review, we identify the available literature regarding the pharmacological augmentation to AD in TRD. Research in the main psychiatric databases was performed (PubMed, ISI Web of Knowledge, PsychInfo). Only original articles in English with the main topic being pharmacological augmentation in TRD and presenting a precise definition of TRD were included. Aripiprazole and lithium were the most investigated molecules, and aripiprazole presented the strongest evidence of efficacy. Moreover, olanzapine, quetiapine, cariprazine, risperidone, and ziprasidone showed positive results but to a lesser extent. Brexpiprazole and intranasal esketamine need further study in real-world practice. Intravenous ketamine presented an evincible AD effect in the short-term. The efficacy of adjunctive ADs, antiepileptic drugs, psychostimulants, pramipexole, ropinirole, acetyl-salicylic acid, metyrapone, reserpine, testosterone, T3/T4, naltrexone, SAMe, and zinc cannot be precisely estimated in light of the limited available data. Studies on lamotrigine and pindolol reported negative results. According to our results, aripiprazole and lithium may be considered by clinicians as potential effective augmentative strategies in TRD, although the data regarding lithium are somewhat controversial. Reliable conclusions about the other molecules cannot be drawn. Further controlled comparative studies, standardized in terms of design, doses, and duration of the augmentative treatments, are needed to formulate definitive conclusions.
[本文引用:1]
|
| [15] |
|
| [16] |
Several new antidepressants that inhibit the serotonin (SERT) and norepinephrine transporters (NET) have been introduced into clinical practice the past several years. This report focuses on the further pharmacologic characterization of nefazodone and its metabolites within the serotonergic and noradrenergic systems, in comparison with other antidepressants. By use of radioligand binding assays, we measured the affinity (Ki) of 13 antidepressants and 6 metabolites for the rat and human SERT and NET. The Ki values for eight of the antidepressants and three metabolites were also determined for the rat 5-HT1A, 5-HT2A and muscarinic cholinergic receptors, the guinea pig histamine1 receptor and the human alpha-1 and alpha-2 receptors. These data are useful for predicting side effect profiles and the potential for pharmacodynamic drug-drug interactions of antidepressants. Of particular interest were the findings that paroxetine, generally thought of as a selective SERT antagonist, possesses moderately high affinity for the NET and that venlafaxine, which has been described as a "dual uptake inhibitor", possesses weak affinity for the NET. We observed significant correlations in SERT (r = 0.965) or NET (r = 0.983) affinity between rat and human transporters. Significant correlations were also observed between muscarinic cholinergic and NET affinity. There are several significant correlations between affinities for the 5-HT1A, 5-HT2A, histamine1, alpha-1 and alpha-2 receptors. These novel findings, not widely described previously, suggest that many of the individual drugs studied in these experiments possess some structural characteristic that determines affinity for several G protein-coupled, but not muscarinic, receptors.
PMID:9400006
[本文引用:2]
|
| [17] |
|
| [18] |
|
| [19] |
Metabolic reactions that occur at alkylamino moieties may provide insight into the roles of these moieties when they are parts of drug molecules that act at different receptors. N-dealkylation of N,N-dialkylamino moieties has been associated with retaining, attenuation or loss of pharmacologic activities of metabolites compared to their parent drugs. Further, N-dealkylation has resulted in clinically used drugs, activation of prodrugs, change of receptor selectivity, and providing potential for developing fully-fledged drugs. While both secondary and tertiary alkylamino moieties (open chain aliphatic or heterocyclic) are metabolized by CYP450 isozymes oxidative N-dealkylation, only tertiary alkylamino moieties are subject to metabolic N-oxidation by Flavin-containing monooxygenase (FMO) to give N-oxide products. In this review, two aspects will be examined after surveying the metabolism of representative alkylamino-moieties-containing drugs that act at various receptors (i) the pharmacologic activities and relevant physicochemical properties (basicity and polarity) of the metabolites with respect to their parent drugs and (ii) the role of alkylamino moieties on the molecular docking of drugs in receptors. Such information is illuminative in structure-based drug design considering that fully-fledged metabolite drugs and metabolite prodrugs have been, respectively, developed from N-desalkyl and N-oxide metabolites.
[本文引用:1]
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
Schizophrenia is a severe psychotic disorder that is diagnosed by the presence of hallucinations or delusions along with disorganized speech, disorganized thought, or negative symptoms that are present for at least six months. Roughly 1 in 10,000 people a year are diagnosed with this psychiatric disorder. It is a chronic disorder requiring a lifetime of treatment of which antipsychotics have been the mainstay of this treatment. First-generation antipsychotics have dystonia, parkinsonism, and development of Tardive Dyskinesia as major side effects, and they are also nonspecific in terms of their actions. Second Generation antipsychotics target more specific dopamine and sometimes serotonin receptors with less dystonic side effects; however, there are additional concerns for the development of metabolic syndrome. This review aims to look at new medication on the market, lumateperone, for the treatment of Schizophrenia. In one four week study with 60mg and 120mg of Lumateperone compared, 4mg of Risperdal, and a placebo found that Lumateperone significantly decreased the total Positive and Negative Syndrome Scale (PANSS) from baseline. Safety analysis of this study also found that Lumateperone was not associated with EPS or significant weight gain. Another study found that 42mg of Lumateperone significantly decreased PANSS score over placebo and 28mg of Lumateperone with associated TEAEs of somnolence, sedation, fatigue, and constipation. In an open-label safety, patients were switched from their current antipsychotic to Lumateperone and then switched back to their previous treatment after six weeks. PATIENTS were found to have statistically significant improvements in metabolic parameters, weight, and endocrine parameters, which were all lost when they were switched back to their previous treatment and their schizophrenic symptoms at pre-trial levels or improved them while on Lumateperone. In a continuation of the previous study over 12 months, 4 TEAEs occurred in 5% or more of the participants: diarrhea, dry mouth, weight decrease, and headache. Prolactin, metabolic labs, BMI, and weight all decreased as compared to the standard of care. Pooled studies revealed EPS related TEAEs were less frequent in patients receiving 42 mg lumateperone over Risperdal. Another pooled study looked at the safety profile; they found patients treated with lumateperone, two TEAEs occurred at twice the placebo rate and at a rate of 5% or more: dry mouth (5% vs. 2.2%) and sedation (24.1% vs. 10.0%) though TEAE discontinuation rates were lower than with Risperdal. Taken together, data from these trials suggest that lumateperone can effectively treat positive symptoms, negative symptoms, and cognitive dysfunction in schizophrenia. Lumateperone entrance to the market introduces an innovative way to treat schizophrenia featuring both a novel mechanism of action and a markedly reduced side effect profile. Further research is needed to determine the efficacy of Lumateperone in treating bipolar disorder in addition to schizophrenia. Copyright © 1964–2019 by MedWorks Media Inc, Los Angeles, CA All rights reserved. Printed in the United States.
PMID:33012872
[本文引用:1]
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
Atrophy of neurons in the prefrontal cortex (PFC) plays a key role in the pathophysiology of depression and related disorders. The ability to promote both structural and functional plasticity in the PFC has been hypothesized to underlie the fast-acting antidepressant properties of the dissociative anesthetic ketamine. Here, we report that, like ketamine, serotonergic psychedelics are capable of robustly increasing neuritogenesis and/or spinogenesis both in vitro and in vivo. These changes in neuronal structure are accompanied by increased synapse number and function, as measured by fluorescence microscopy and electrophysiology. The structural changes induced by psychedelics appear to result from stimulation of the TrkB, mTOR, and 5-HT2A signaling pathways and could possibly explain the clinical effectiveness of these compounds. Our results underscore the therapeutic potential of psychedelics and, importantly, identify several lead scaffolds for medicinal chemistry efforts focused on developing plasticity-promoting compounds as safe, effective, and fast-acting treatments for depression and related disorders. Copyright © 2018 The Author(s). Published by Elsevier Inc. All rights reserved.
DOI:S2211-1247(18)30755-1
PMID:29898390
[本文引用:2]
|
| [43] |
|
| [44] |
Psilocybin is a naturally occurring alkaloid, pharmacologically similar to the classic hallucinogen lysergic acid diethylamide (LSD). Although primarily used as a recreational drug or an entheogen in particular cultural settings, recent population based studies have shown that it does not lead to serious physical or mental health problems or dependent use. In view of recent work demonstrating psilocybin’s potential to increase subjective sense of wellbeing and because of its novel mechanism of 5-HT2A serotonin receptor agonism, it is being explored for possible therapeutic utility in mood and anxiety disorders.
[本文引用:1]
|
| [45] |
Psilocybin has shown promise as a treatment for depression but its therapeutic mechanisms are not properly understood. In contrast to the presumed actions of antidepressants, we recently found increased amygdala responsiveness to fearful faces one day after open-label treatment with psilocybin (25 mg) in 19 patients with treatment-resistant depression, which correlated with treatment efficacy.
[本文引用:1]
|
| [46] |
|
| [47] |
|
| [48] |
Drugs that target the human serotonin 2A receptor (5-HTR) are used to treat neuropsychiatric diseases; however, many have hallucinogenic effects, hampering their use. Here, we present structures of 5-HTR complexed with the psychedelic drugs psilocin (the active metabolite of psilocybin) and d-lysergic acid diethylamide (LSD), as well as the endogenous neurotransmitter serotonin and the nonhallucinogenic psychedelic analog lisuride. Serotonin and psilocin display a second binding mode in addition to the canonical mode, which enabled the design of the psychedelic IHCH-7113 (a substructure of antipsychotic lumateperone) and several 5-HTR β-arrestin-biased agonists that displayed antidepressant-like activity in mice but without hallucinogenic effects. The 5-HTR complex structures presented herein and the resulting insights provide a solid foundation for the structure-based design of safe and effective nonhallucinogenic psychedelic analogs with therapeutic effects.
DOI:10.1126/science.abl8615
PMID:35084960
[本文引用:1]
|

{kind=link}
{kind=link}