中国科技论文统计源期刊 中文核心期刊
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖
美国《化学文摘》《国际药学文摘》
《乌利希期刊指南》
WHO《西太平洋地区医学索引》来源期刊
日本科学技术振兴机构数据库(JST)
第七届湖北十大名刊提名奖

 , 杨志勇
, YANG Zhiyong
, 杨志勇
, YANG Zhiyong
目的 建立甲硝唑原料药有关物质的高效液相色谱测定方法和杂质谱。方法 采用Welch Ultimate®XB-C18色谱柱(4.6 mm×250 mm,5 μm);以甲醇-1.36 g·L-1磷酸二氢钾溶液(20∶80)为流动相;流速1 mL·min-1;检测波长:315 nm,主成分自身对照法测定有关物质。结果 甲硝唑与有关物质具有良好的分离度,对近6年生产的20批样品进行了测定,均满足质量控制标准。杂质谱的研究能有效监控甲硝唑合成工艺和杂质变化情况。结论 该方法简便、快速、灵敏,能有效控制甲硝唑原料药中有关物质,同时杂质谱研究有助于保证甲硝唑质量稳定性检测。
Objective To establish HPLC determination method and impurity profile of the related substances in metronidazole. Methods A Welch Ultimate®XB-C18(4.6 mm×250 mm, 5 μm)was used with a mobile phase consisting of methanol-1.36 g·L-1 solution of potassium dihydrogen phosphate (20∶80). The detection wavelength was 315 nm and the flow rate was 1 mL·min-1. Its related substances were determined by principal component self-contrast method. Results Good separation of metronidazole and the impurities could be achieved. Twenty batches of samples in the past six years were determined which meet quality standards. The study of impurity profiles could effectively monitor the synthetic process and the change of impurities in metronidazole.Conclusion The method is simple, quick and sensitive, which can be used to control the related substances in metronidazole. Meanwhile, the impurity profiles ensure the quality stability of metronidazole.
Agilent1100型高效液相色谱仪(美国安捷伦公司),包括紫外检测器,在线脱气机,四元梯度泵,化学工作站; AUW 220D型双量程分析天平(日本岛津公司,感量: 0.1 mg/0.01 mg)。
甲硝唑对照品(中国食品药品检定研究院,批号:100191-200606,含量:100%)。杂质A对照品(批号:100512-200802,含量:99.8%)购于中国食品药品检定研究院,杂质B、C、D、E、F、G对照品(批号分别为1292-014A1、1317-059A8、1317-088A5、1303-099A5、1540-009A3、1319-032A3,含量分别为99.6%,97.0%,99.4%,98.2%,99.9%,99.9%)购自于TLC PharmaChem,其化学结构分别为2-甲基-5-硝基咪唑、4-硝基咪唑、2-(4-硝基-1
色谱柱:Welch Ultimate®XB-C18(4.6 mm×250 mm,5 μm);流动相:甲醇-1.36 g·L-1磷酸二氢钾溶液(20∶80);流速:1 mL·min-1;柱温:40 ℃;检测波长:315 nm;进样体积:10 μL。
供试品溶液:取甲硝唑原料药约50 mg,精密称定,置100 mL量瓶中,用流动相溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液。
对照溶液:精密量取供试品溶液1 mL,置100 mL量瓶,用流动相稀释至刻度,摇匀,精密量取1 mL,置于10 mL量瓶,用流动相稀释至刻度,摇匀,即得。
杂质对照品溶液:分别取甲硝唑对照品及杂质A、B、C、D、E、F、G对照品约5 mg,精密称定,分别置于10 mL量瓶,用流动相溶解并稀释至刻度,摇匀。
系统适用性溶液:取甲硝唑原料药约50 mg精密称定,再分别精密吸取上述杂质A、B、C、D、E、F、G对照品溶液100 μL,置于同一100 mL量瓶,用流动相溶解并稀释至刻度,摇匀,配成含甲硝唑约0.5 mg·mL-1及各杂质对照品0.5 μg·mL-1的溶液。
取空白溶剂、系统适用性溶液和供试品溶液分别进样测定,记录色谱图。系统适用性溶液色谱图中,测得甲硝唑的理论板数>10 000,其他各杂质理论板数>5 000,甲硝唑与前后杂质峰的分离度>2.0,其他各杂质峰分离度均符合要求,实现了杂质A、B、C、D、E、F、G及未知杂质1和未知杂质2等9个杂质的有效分离,保留时间分别为杂质G(3.90 min)、杂质C(4.17 min)、杂质B(4.47 min)、杂质E(4.86 min)、未知杂质1(5.10 min)、未知杂质2(5.37 min)、杂质A(5.68 min)、杂质D(6.19 min)、杂质F(13.06 min),见
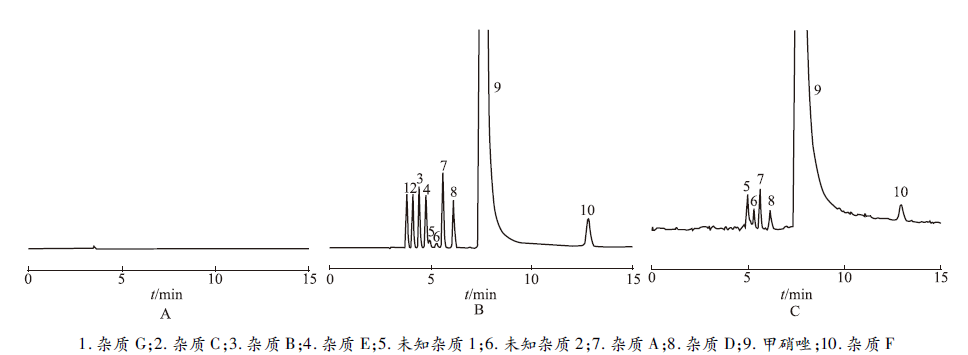
图1
空白溶剂(A)、系统适用性溶液(B)和供试品溶液(C)高效液相色谱图
1.impurity G ; 2.impurity C; 3.impurity B; 4.impurity E; 5.unknown impurity 1; 6.unknown impurity 2; 7.impurity A; 8.impurity D; 9.metronidazole; 10.impurity F
Fig.1 HPLC chromatograms of blank solvent(A), system suitability solution(B)and test solution(C)
取甲硝唑原料药50 mg精密称定,5份,分别置100 mL量瓶,进行酸破坏(加1 mol·L-1盐酸溶液10 mL,室温下避光放置6 h,用1 mol·L-1氢氧化钠溶液调pH至中性)、碱破坏(加0.8 mol·L-1氢氧化钠溶液10 mL,室温下避光放置1 h,用0.8 mol·L-1盐酸溶液调pH至中性)、氧化破坏(加30%过氧化氢溶液20 mL,室温下放置1 d)、高温破坏(加适量水使溶解,95 ℃水浴加热1 d)、光照破坏[加适量水使溶解,(4 500±500) lx,1 d]。破坏结束后,用流动相稀释至刻度。按“2. 1”项色谱条件测定。结果表明,在选定的色谱条件下,经酸、碱破坏后有降解,其中碱破坏最为明显,氧化破坏次之,酸、光照、高温破坏后降解不明显。各降解产物峰与主峰分离度均符合要求,见
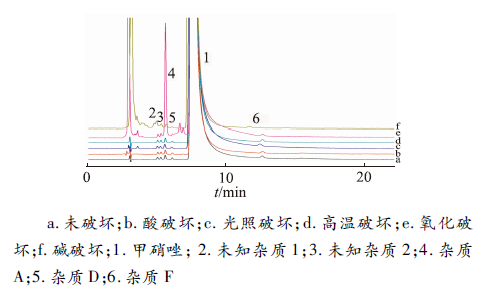
图2
方法专属性考察色谱图
a. undestroyed; b. destroyed by acid; c. destroyed by intense light; d. destroyed by excessive heating; e. destroyed by oxidation; f. destroyed by alkali; 1. metronidazole; 2. unknown impurity 1; 3. unknown impurity 2; 4. impurity A; 5. impurity D; 6. impurity F
Fig.2 HPLC chromatograms of the specificity test
精密吸取甲硝唑对照品溶液及各杂质对照品溶液各500 μL,置同一100 mL量瓶,加流动相稀释至刻度,摇匀,即得混合对照品溶液,再分别量取0.5,1.0,5.0,10.0,20.0,40.0 mL置于50 mL量瓶,加流动相稀释至刻度,摇匀,得到系列标准曲线溶液。按“2.1”项色谱条件,进样,记录色谱图。以峰面积值为纵坐标(
表1 甲硝唑及有关物质的线性关系和检测限
Tab.1 Linearity and limit of detection (LOD) of metronidazole and related substances
取线性关系考察项下混合对照品溶液,将其稀释至一定浓度,以3倍噪声比(
精密吸取甲硝唑对照品溶液及各杂质对照品溶液各100 μL,置于同一100 mL量瓶,加流动相稀释至刻度,摇匀,按“2.1”项色谱条件平行进样5次,计算各色谱峰的面积。甲硝唑及杂质A、B、C、D、E、F、G的RSD分别为0.55%,0.43%,0.76%,0.34%,0.61%,0.49%,0.63%,0.32%。RSD均小于0.8%,仪器精密度良好。
取甲硝唑样品平行制备6份供试品溶液,按“2.1”项色谱条件进样测定,计算供试品溶液中各色谱峰的面积,甲硝唑及杂质A、D、F,未知杂质1、2的RSD分别为1.27%,10.66%,11.46%,3.01%,8.88%,6.87%。甲硝唑RSD<2.0%,由于供试品溶液杂质含量极低,仅在定量限附近,仅需满足RSD<10%,因各杂质RSD在3.01%~11.46%之间,表明仪器重复性良好。
取甲硝唑样品,按“2.2”项制备供试品溶液,室温放置,分别在第0,2,4,6,8,10,12 h进样测定,测得甲硝唑及杂质A、D、F,未知杂质1、2的峰面积RSD分别为1.09 %,4.54%,9.15%,12.16%,10.48%,6.61%。甲硝唑溶液在室温下放置12 h峰面积RSD为1.09%,稳定性好。虽然杂质在溶液中放置12 h后峰面积的RSD为4.5%~12.2%,但由于杂质含量极低,仅在定量限附近,只需满足RSD<20%的要求,稳定性较好。
按 “2.2”项溶液制备方法配制20批甲硝唑原料药供试品溶液,进样测定,记录3倍主峰保留时间,以主成分自身对照法计算各杂质含量,结果见
表2 甲硝唑有关物质测定结果
Tab.2 Results of metronidazole related substances %
从测定结果可知,未知杂质1和未知杂质2的含量在0.00%~0.02%之间。由于甲硝唑每日服用最大剂量>2 g,根据原料药杂质限度规定,若未知杂质含量未达报告限度(0.03%)、鉴定限度(0.05%)及质控限度(0.05%)可不对其进行报告、结构鉴定和质量控制,因此,目前可不对未知杂质1和杂质2进行结构确证。杂质A、D、F的含量在0.00%~0.04%,0.00%~0.01%, 0.00%~0.03%之间,其他杂质未检出,总杂质含量在0.00%~0.09%之间。表明甲硝唑原料药性质稳定,在室温避光下储存1~5年的原料药样品未出现新的降解杂质,已知杂质和未知杂质均在限度以内。
参考《欧洲药典》 [4],结合实验结果,将甲硝唑中杂质限度定为:总杂质含量≤0.2%,单个未知杂质含量≤0.05%,单个已知杂质含量≤0.1%,含量<0.01%的杂质忽略不计。
2010年版《中华人民共和国药典》选择甲醇-水作为流动相用于检测甲硝唑有关物质,按此色谱条件进样发现,系统适用性溶液色谱图中杂质G未出峰,而为有效检出所有杂质,加入磷酸二氢钾能够有效抑制杂质G的解离,使其出峰,以甲醇-磷酸二氢钾缓冲盐作为流动相更合理。笔者在本实验考察了C18和C8柱及不同厂家C18柱对实验中甲硝唑和各杂质分离的影响,甲硝唑样品的测定不受C18和C8柱的影响,但C8柱不能将系统适用性溶液中杂质G、C、B、E及未知杂质有效分离。由于杂质B、C、E、G结构相近,在色谱柱中的保留行为接近,将四者同时分离具有一定的难度,因此,选择合适的C18柱对甲硝唑原料药有关物质的检查十分重要。
甲硝唑为2-甲基-5-硝基咪唑与环氧乙烷羟乙基化所得。杂质A为起始原料2-甲基-5-硝基咪唑。经有关物质检查,杂质B为起始原料中有关物质。杂质C、D、E、F、G分别为合成反应中的副产物。杂质C和杂质D是杂质B与环氧乙烷反应的副产物,杂质F为甲硝唑与环氧乙烷反应的副产物,杂质G为甲硝唑结构中羟基氧化为羧基所得。原料或副产物在反应过程中还可能发生其他氧化反应或与环氧乙烷的反应,以及在储存和运输中可能发生的降解反应,都是未知杂质产生的可能原因。
通过对比同一厂家不同时期生产的甲硝唑中杂质的种类和数量的关系,可了解厂家在不同阶段工艺改进与参数优化方面的变更情况;而对近期连续生产的甲硝唑杂质谱进行种类及数量的分布统计,可动态反映当前阶段生产工艺控制水平;同时,也可与其他厂家生产的甲硝唑原料药杂质谱比较,不仅反映产品质量在杂质控制水平上的差异,还可反映甲硝唑在合成工艺上的差异性。
对稳定性实验研究建立的杂质谱可定义为稳定性杂质谱。通过对比实验样品与空白实验样品的杂质谱,可分析哪些杂质是由工艺生产带进(此类杂质在稳定性实验中含量一般无显著变化),哪些杂质是由降解产生的(此类杂质在稳定性实验中含量可能显著改变)。通过对稳定性杂质谱进行分析,可了解杂质的基本组成及产生原因,杂质的变化情况,为制剂中杂质的定性、定量研究提供实验准备。
甲硝唑杂质谱的研究是药品质量研究、质量控制和安全性研究的基础。
本实验色谱条件将甲硝唑及生产过程中可能产生的9种有关物质进行了有效分离,方法简便、灵敏,对甲硝唑原料药单个杂质和总杂质进行了有效地控制。检出的已知杂质A为合成甲硝唑的起始原料,已知杂质D、F是合成的副产物。甲硝唑杂质谱的研究能够对原料药的合成工艺及杂质变化情况进行有效监测,保证其质量稳定性。虽然未知杂质均未达到报告限度,为了提高原料药的质量控制,未知杂质的定性还需要进一步研究。
The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}